

Genome-wide prediction of disease variant effects with a deep protein language model
- PMID: 37563329
- PMCID: PMC10484790
- DOI: 10.1038/s41588-023-01465-0
Genome-wide prediction of disease variant effects with a deep protein language model
Abstract
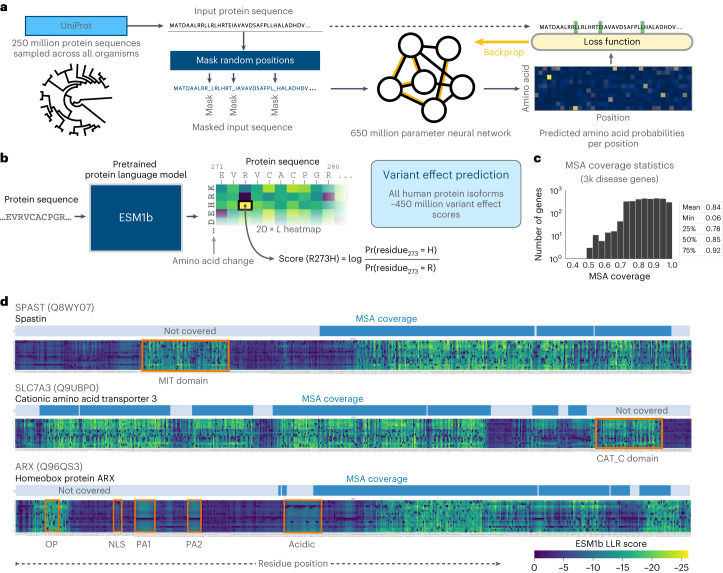
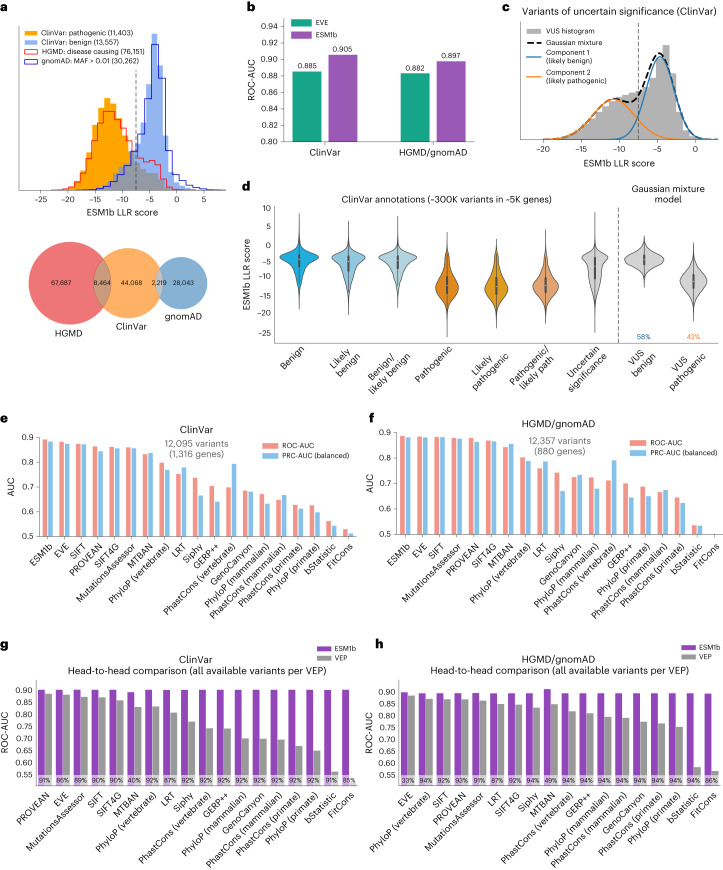
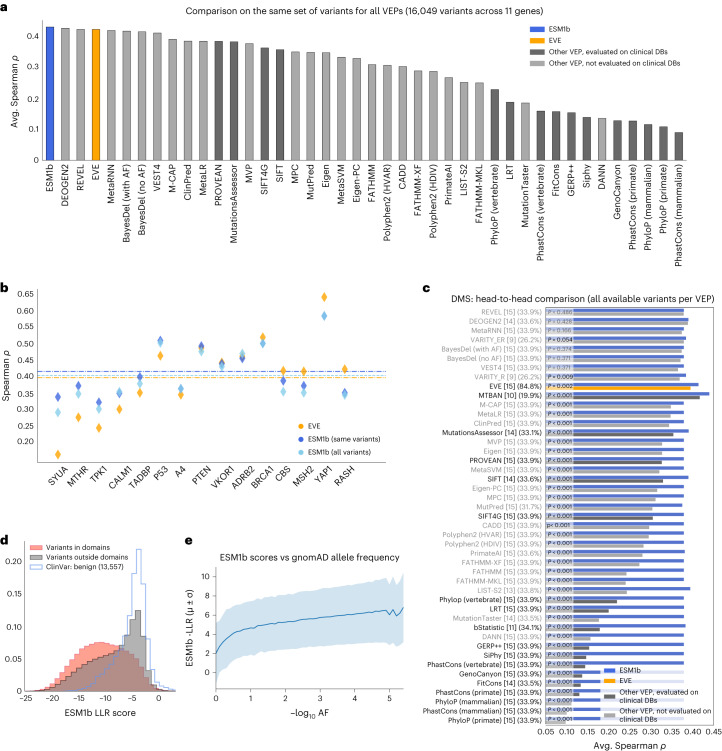
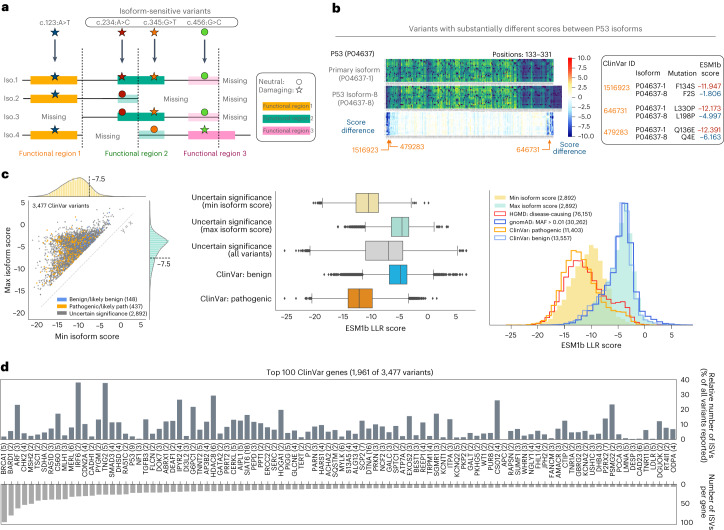
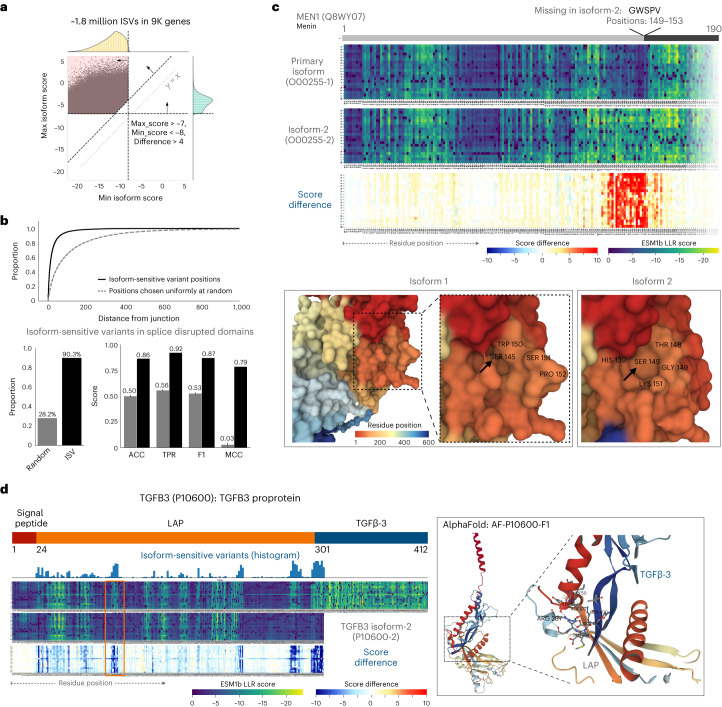
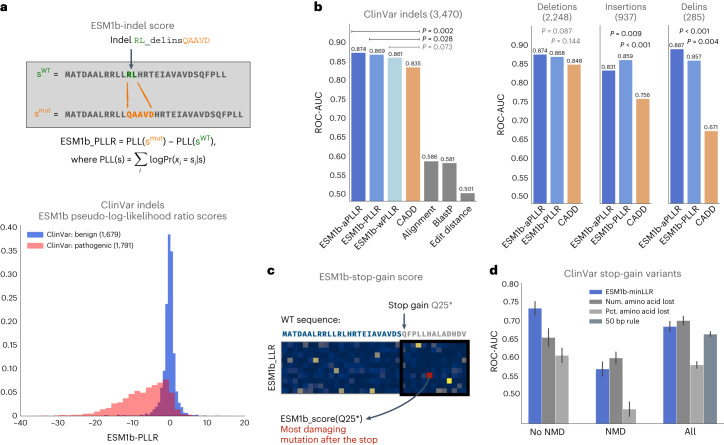
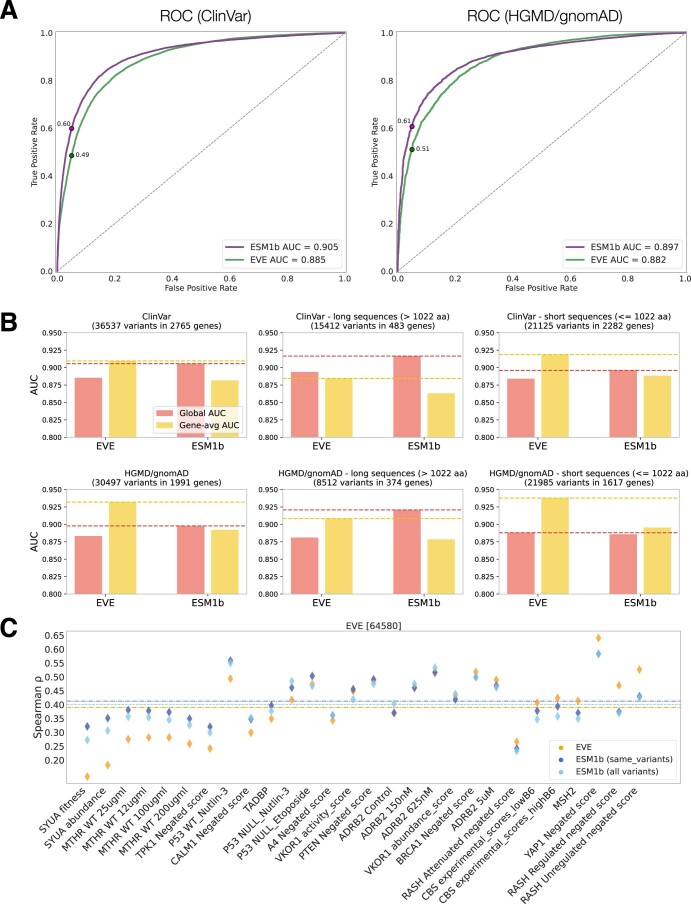
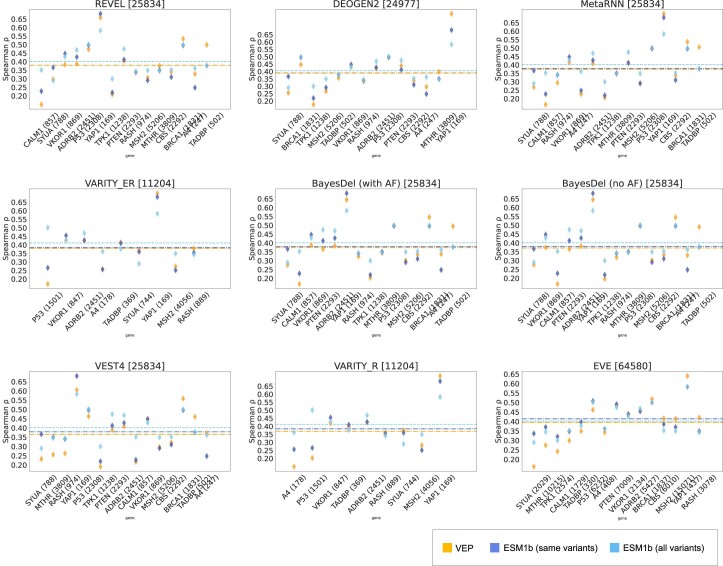
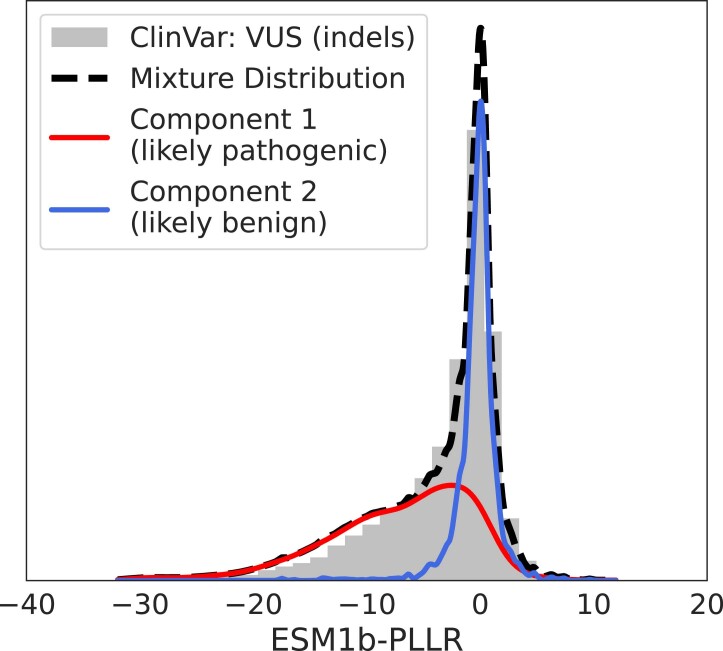
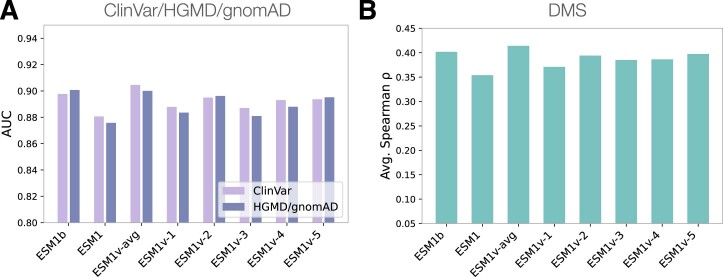
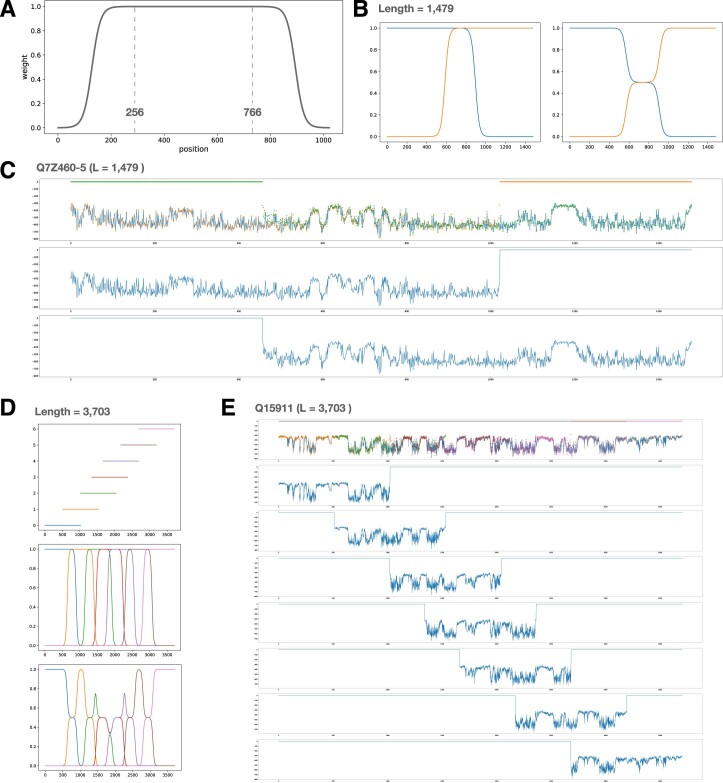
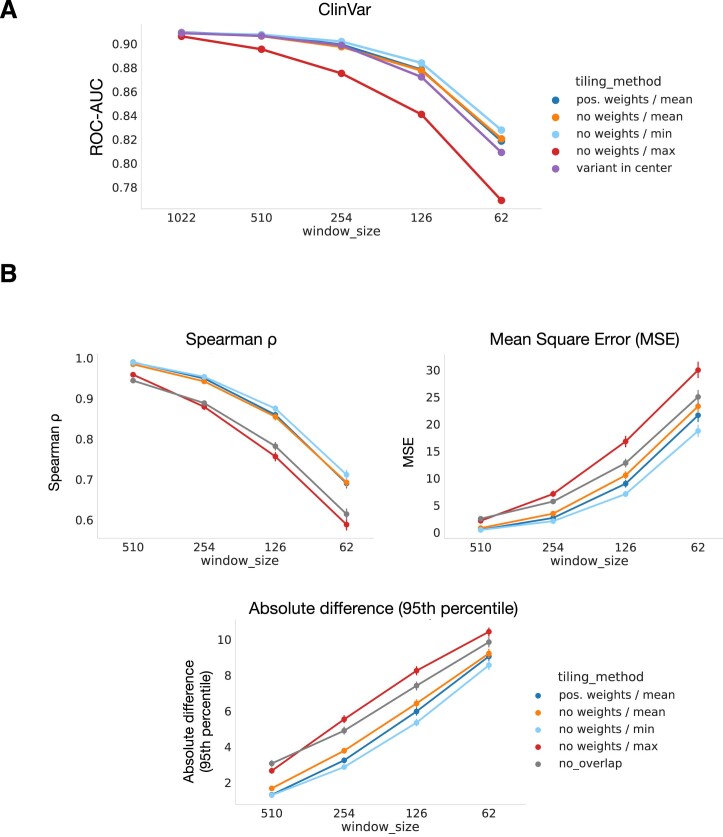
Predicting the effects of coding variants is a major challenge. While recent deep-learning models have improved variant effect prediction accuracy, they cannot analyze all coding variants due to dependency on close homologs or software limitations. Here we developed a workflow using ESM1b, a 650-million-parameter protein language model, to predict all ~450 million possible missense variant effects in the human genome, and made all predictions available on a web portal. ESM1b outperformed existing methods in classifying ~150,000 ClinVar/HGMD missense variants as pathogenic or benign and predicting measurements across 28 deep mutational scan datasets. We further annotated ~2 million variants as damaging only in specific protein isoforms, demonstrating the importance of considering all isoforms when predicting variant effects. Our approach also generalizes to more complex coding variants such as in-frame indels and stop-gains. Together, these results establish protein language models as an effective, accurate and general approach to predicting variant effects.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
