

Computational and Experimental Analyses for Pathogenicity Prediction of ACVRL1 Missense Variants in Hereditary Hemorrhagic Telangiectasia
- PMID: 37568404
- PMCID: PMC10419700
- DOI: 10.3390/jcm12155002
Computational and Experimental Analyses for Pathogenicity Prediction of ACVRL1 Missense Variants in Hereditary Hemorrhagic Telangiectasia
Abstract
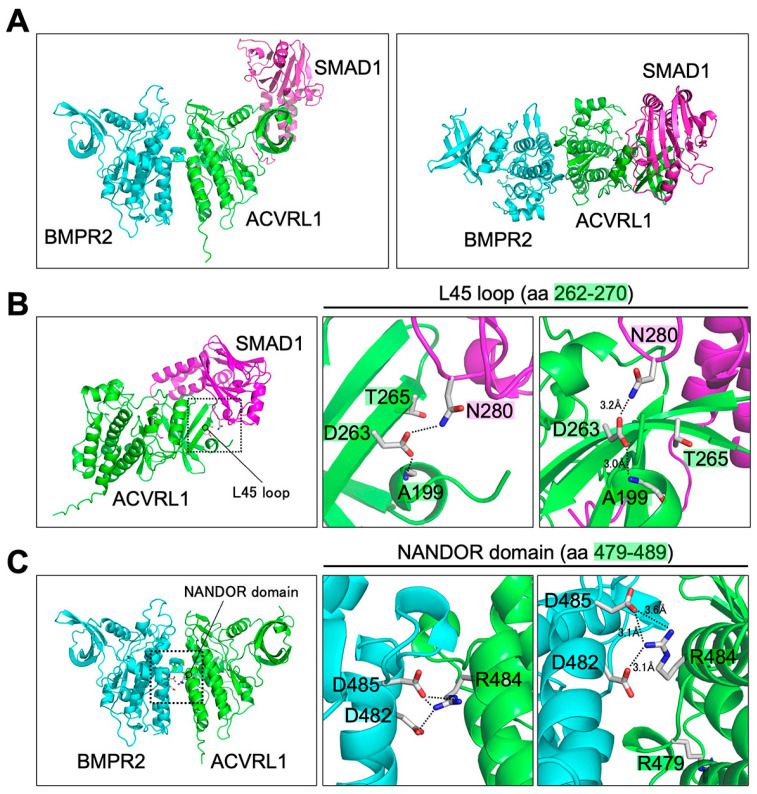
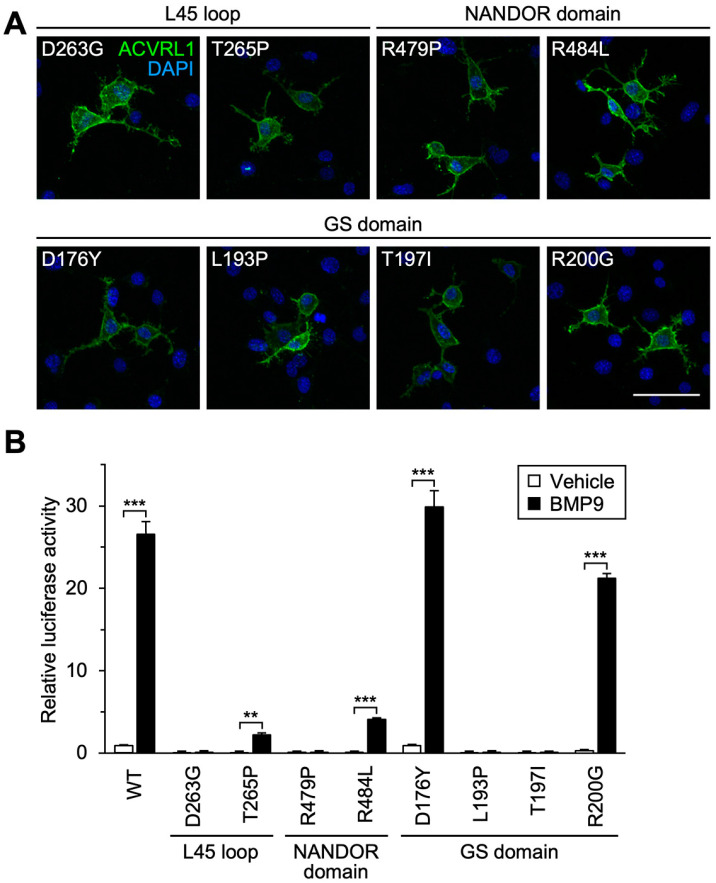
Hereditary hemorrhagic telangiectasia (HHT) is a vascular disease caused by the defects of ALK1/ACVRL1 receptor signaling. In this study, we evaluated 25 recently identified ACVRL1 missense variants using multiple computational pathogenicity classifiers and experimentally characterized their signal transduction capacity. Three extracellular residue variants showed no detectable cell surface expression and impairment of bone morphogenetic protein 9 (BMP9) responsiveness of SMAD-dependent transcription in luciferase assays. Four variants with amino acid replacement in the motifs essential for the intracellular kinase function lost SMAD-dependent signaling. Most of other variations in the kinase domain also caused marked downregulation of signaling; however, two variants behaved as the wild-type ACVRL1 did, while computational classifiers predicted their functional abnormalities. Three-dimensional structure prediction using the ColabFold program supported the significance of the L45 loop and NANDOR domain of ACVRL1 for its association with SMAD1 and BMPR2, respectively, and the variations in these motifs resulted in the reduction of SMAD signaling. On the other hand, two of the GS domain variants maintained high signal transduction capacity, which did not accord with their computational pathogenicity prediction. These results affirm the requirement of a combinatory approach using computational and experimental analyses to accurately predict the pathogenicity of ACVRL1 missense variants in the HHT patients.
Keywords: ACVRL1; ALK1; BMP; SMAD signaling; hereditary hemorrhagic telangiectasia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Shovlin C.L., Guttmacher A.E., Buscarini E., Faughnan M.E., Hyland R.H., Westermann C.J., Kjeldsen A.D., Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am. J. Med. Genet. 2000;91:66–67. doi: 10.1002/(SICI)1096-8628(20000306)91:1<66::AID-AJMG12>3.0.CO;2-P. - DOI - PubMed
-
- Oh S.P., Seki T., Goss K.A., Imamura T., Yi Y., Donahoe P.K., Li L., Miyazono K., ten Dijke P., Kim S., et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
