

An information-theoretic approach to single cell sequencing analysis
- PMID: 37573291
- PMCID: PMC10422744
- DOI: 10.1186/s12859-023-05424-8
An information-theoretic approach to single cell sequencing analysis
Abstract
Background: Single-cell sequencing (sc-Seq) experiments are producing increasingly large data sets. However, large data sets do not necessarily contain large amounts of information.
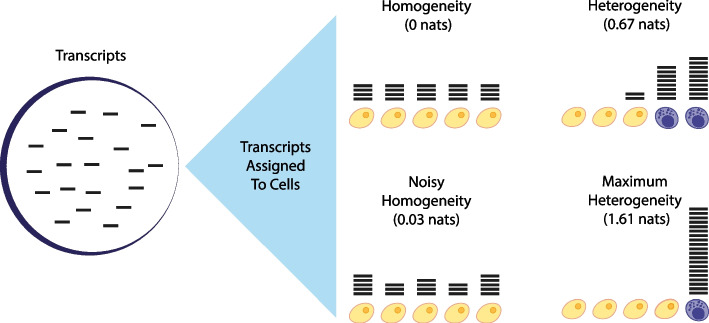
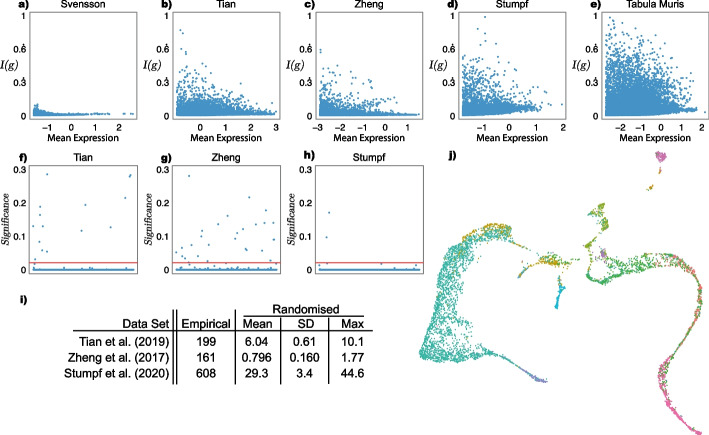
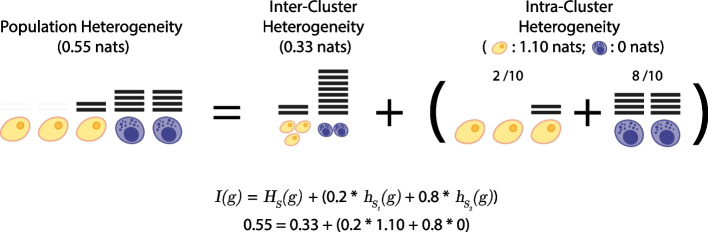
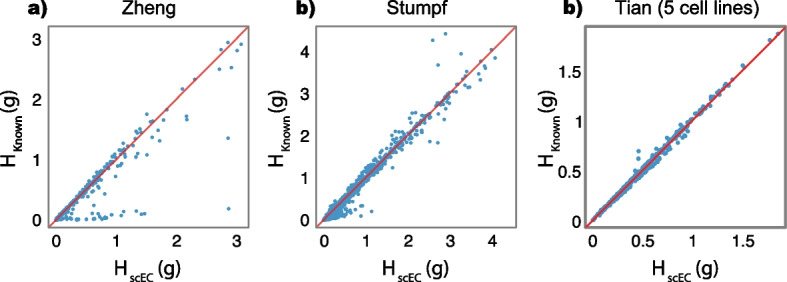
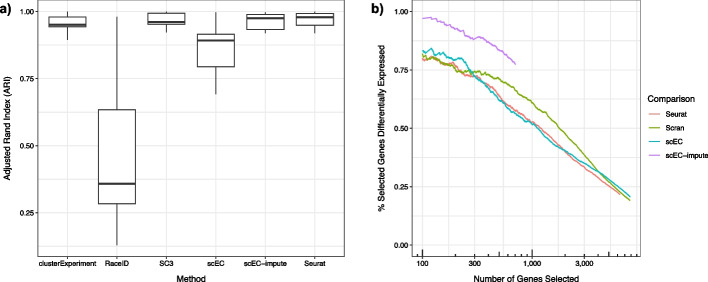
Results: Here, we formally quantify the information obtained from a sc-Seq experiment and show that it corresponds to an intuitive notion of gene expression heterogeneity. We demonstrate a natural relation between our notion of heterogeneity and that of cell type, decomposing heterogeneity into that component attributable to differential expression between cell types (inter-cluster heterogeneity) and that remaining (intra-cluster heterogeneity). We test our definition of heterogeneity as the objective function of a clustering algorithm, and show that it is a useful descriptor for gene expression patterns associated with different cell types.
Conclusions: Thus, our definition of gene heterogeneity leads to a biologically meaningful notion of cell type, as groups of cells that are statistically equivalent with respect to their patterns of gene expression. Our measure of heterogeneity, and its decomposition into inter- and intra-cluster, is non-parametric, intrinsic, unbiased, and requires no additional assumptions about expression patterns. Based on this theory, we develop an efficient method for the automatic unsupervised clustering of cells from sc-Seq data, and provide an R package implementation.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc: Ser B (Methodol) 1995;57(1):289–300.
-
- Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech: Theory Exp. 2008;2008(10):P10008. doi: 10.1088/1742-5468/2008/10/P10008. - DOI
-
- Byrd RH, Lu P, Nocedal J, Zhu C. A limited memory algorithm for bound constrained optimization. SIAM J Sci Comput. 1995;16(5):1190–1208. doi: 10.1137/0916069. - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
