

Characterizing cancer metabolism from bulk and single-cell RNA-seq data using METAFlux
- PMID: 37573313
- PMCID: PMC10423258
- DOI: 10.1038/s41467-023-40457-w
Characterizing cancer metabolism from bulk and single-cell RNA-seq data using METAFlux
Abstract
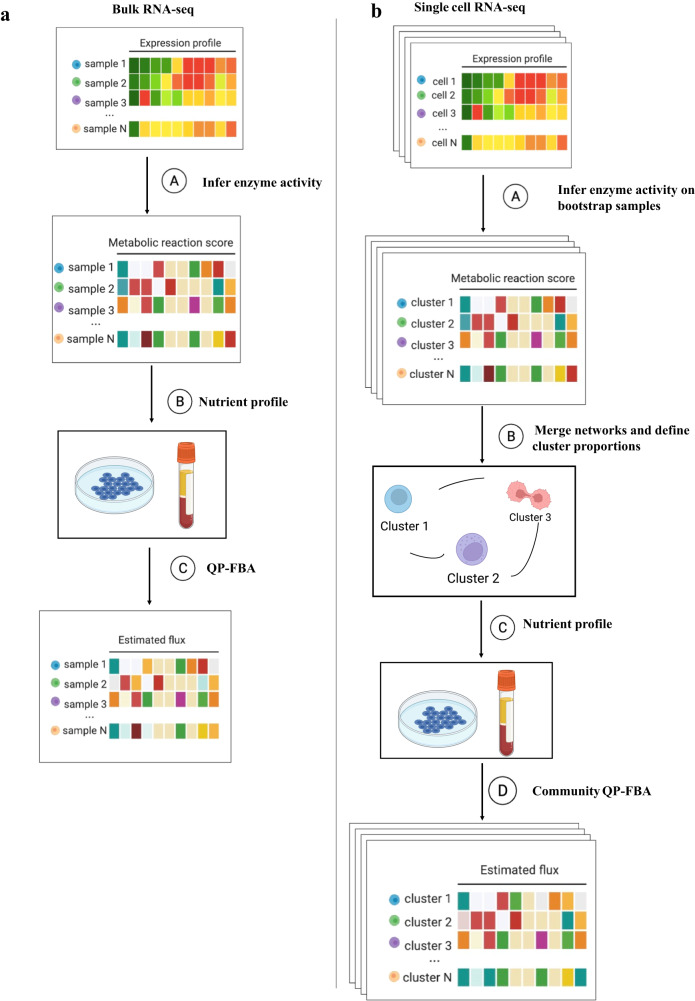
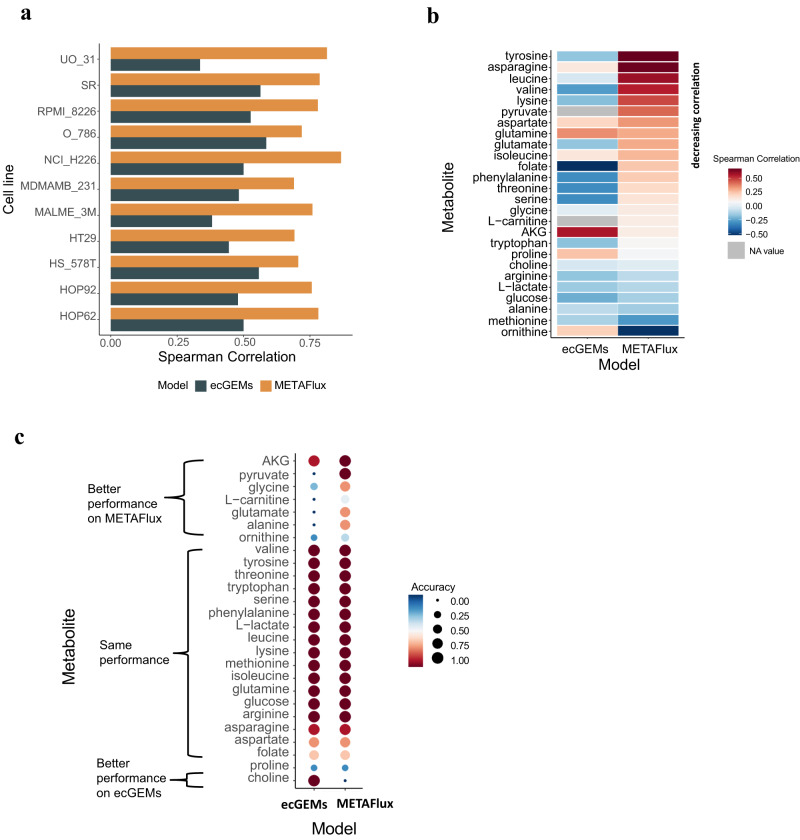
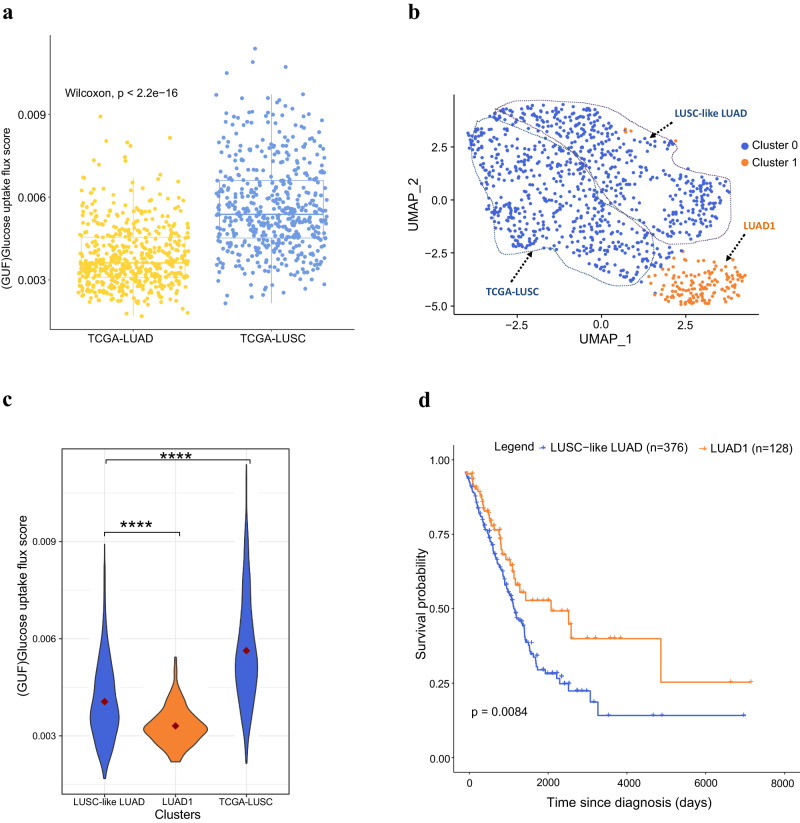
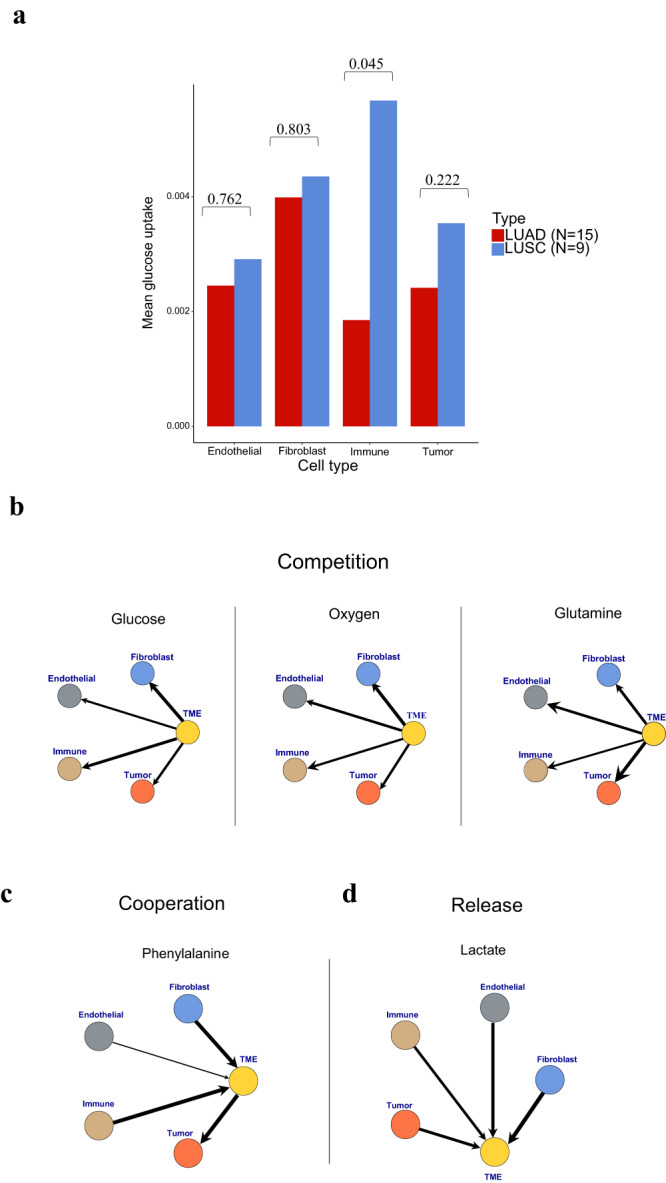
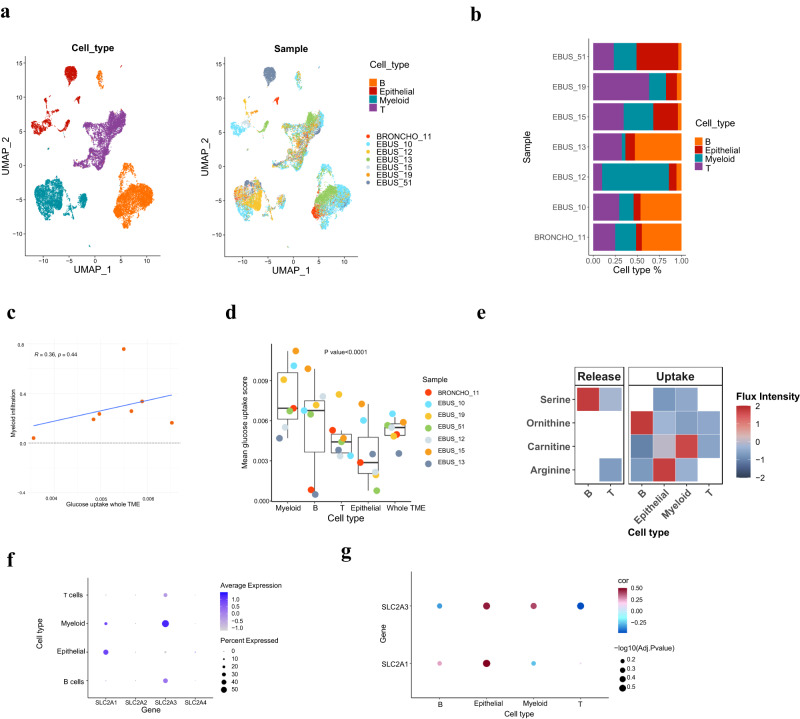
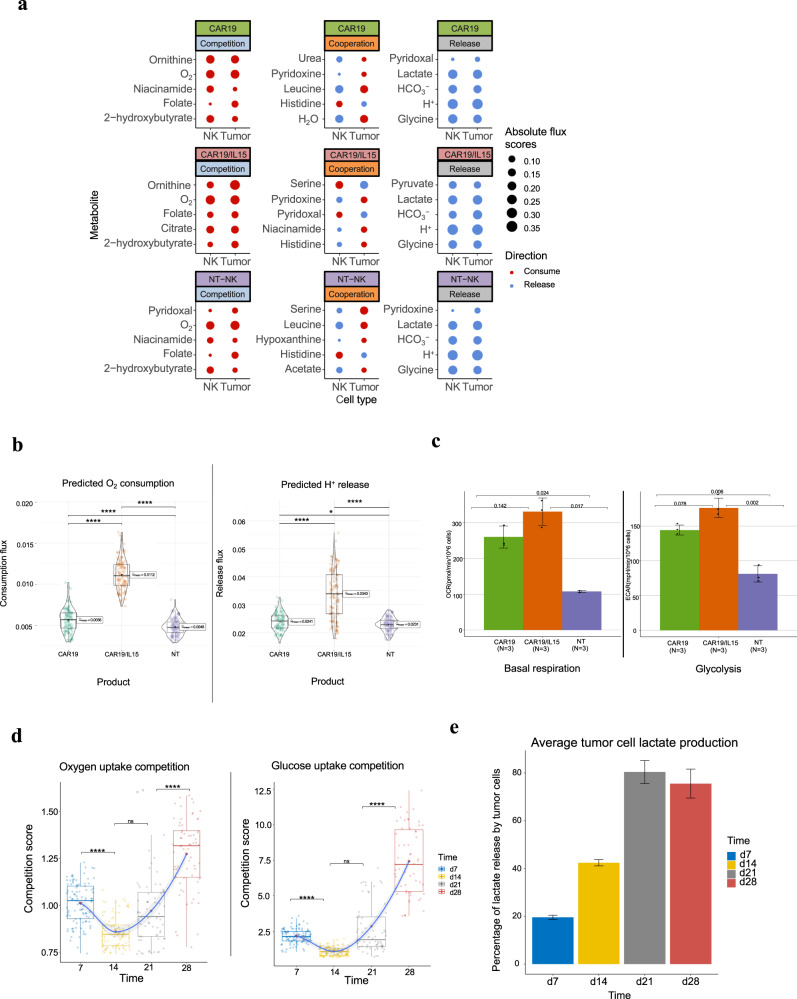
Cells often alter metabolic strategies under nutrient-deprived conditions to support their survival and growth. Characterizing metabolic reprogramming in the tumor microenvironment (TME) is of emerging importance in cancer research and patient care. However, recent technologies only measure a subset of metabolites and cannot provide in situ measurements. Computational methods such as flux balance analysis (FBA) have been developed to estimate metabolic flux from bulk RNA-seq data and can potentially be extended to single-cell RNA-seq (scRNA-seq) data. However, it is unclear how reliable current methods are, particularly in TME characterization. Here, we present a computational framework METAFlux (METAbolic Flux balance analysis) to infer metabolic fluxes from bulk or single-cell transcriptomic data. Large-scale experiments using cell-lines, the cancer genome atlas (TCGA), and scRNA-seq data obtained from diverse cancer and immunotherapeutic contexts, including CAR-NK cell therapy, have validated METAFlux's capability to characterize metabolic heterogeneity and metabolic interaction amongst cell types.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
