

A Novel Pathogenic Variant in the MN1 Gene in a Patient Presenting with Rhombencephalosynapsis and Craniofacial Anomalies, Expanding MN1 C-terminal Truncation Syndrome
- PMID: 37575653
- PMCID: PMC10421676
- DOI: 10.1055/s-0041-1728650
A Novel Pathogenic Variant in the MN1 Gene in a Patient Presenting with Rhombencephalosynapsis and Craniofacial Anomalies, Expanding MN1 C-terminal Truncation Syndrome
Abstract
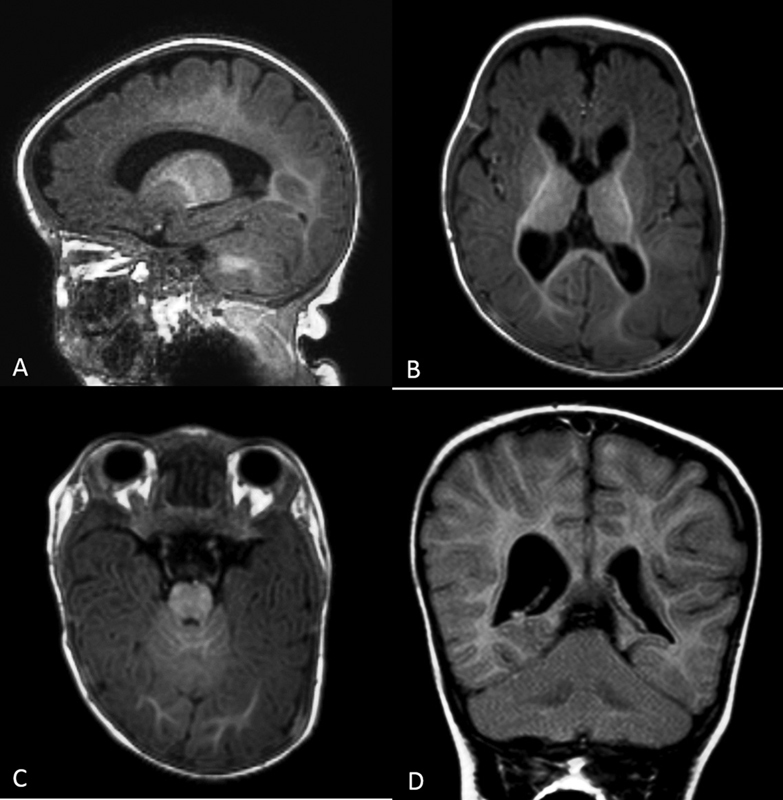
Meningioma-1 is a transcription activator that regulates mammalian palate development and is required for appropriate osteoblast proliferation, motility, differentiation, and function. Microdeletions involving the MN1 gene have been linked to syndromes including craniofacial anomalies, such as Toriello-Carey syndrome. Recently, truncating variants in the C-terminal portion of the MN1 transcriptional factor have been linked to a characteristic and distinct phenotype presenting with craniofacial anomalies and partial rhombencephalosynapsis, a rare brain malformation characterized by midline fusion of the cerebellar hemispheres with partial or complete loss of the cerebellar vermis. It has been called MN1 C-terminal truncation (MCTT) syndrome or CEBALID (Craniofacial defects, dysmorphic Ears, Brain Abnormalities, Language delay, and Intellectual Disability) and suggested to be caused by dominantly acting truncated protein MN1 instead of haploinsufficiency. As a proto-oncogene, MN1 is also involved in familial meningioma. In this study, we present a novel case of MCTT syndrome in a female patient presenting with craniofacial anomalies and rhombencephalosynapsis, harboring a de novo pathogenic variant in the MN1 gene: c.3686_3698del, p.(Met1229Argfs*87).
Keywords: MN1; MN1 C-terminal truncation (MCTT) syndrome and craniofacial anomalies; rhombencephalosynapsis.
Thieme. All rights reserved.
Conflict of interest statement
Conflict of Interest None declared.
Figures


References
-
- Lekanne Deprez R H, Riegman P HJ, Groen N A et al. Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene. 1995;10(08):1521–1528. - PubMed
-
- Toriello H V, Carey J C. Corpus callosum agenesis, facial anomalies, Robin sequence, and other anomalies: a new autosomal recessive syndrome? Am J Med Genet. 1988;31(01):17–23. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
