

This is a preprint.
A genetic basis for cancer sex differences revealed in Xp11 translocation renal cell carcinoma
- PMID: 37577497
- PMCID: PMC10418269
- DOI: 10.1101/2023.08.04.552029
A genetic basis for cancer sex differences revealed in Xp11 translocation renal cell carcinoma
Update in
-
A genetic basis for sex differences in Xp11 translocation renal cell carcinoma.Cell. 2024 Oct 3;187(20):5735-5752.e25. doi: 10.1016/j.cell.2024.07.038. Epub 2024 Aug 20. Cell. 2024. PMID: 39168126 Free PMC article.
Abstract
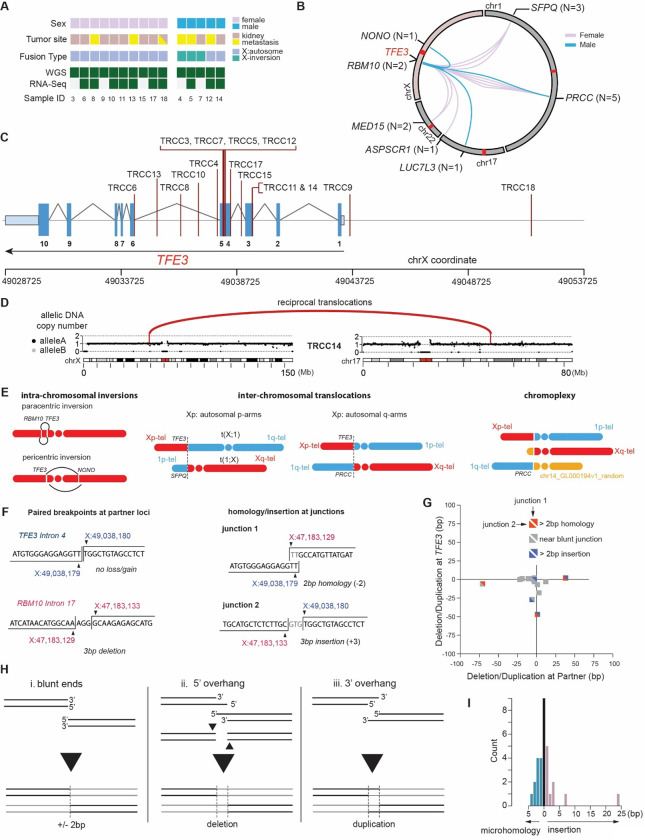
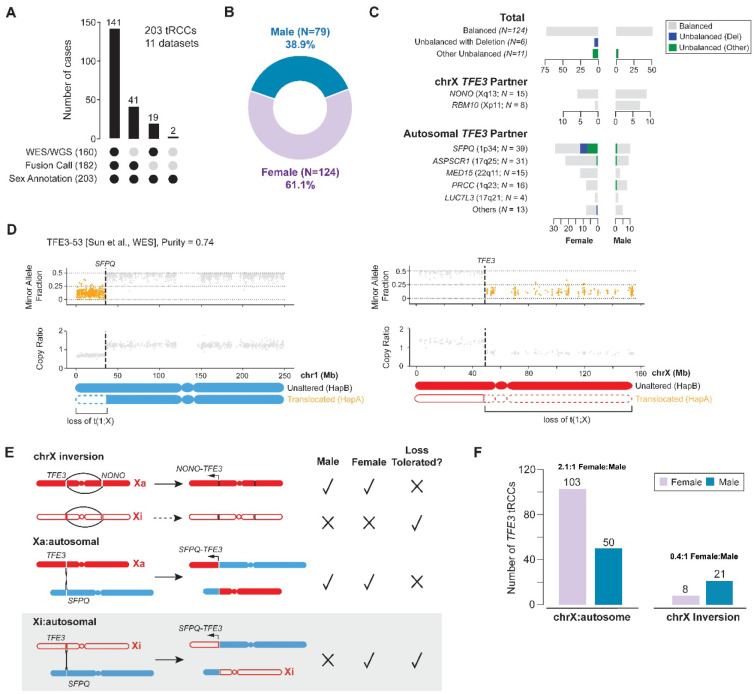
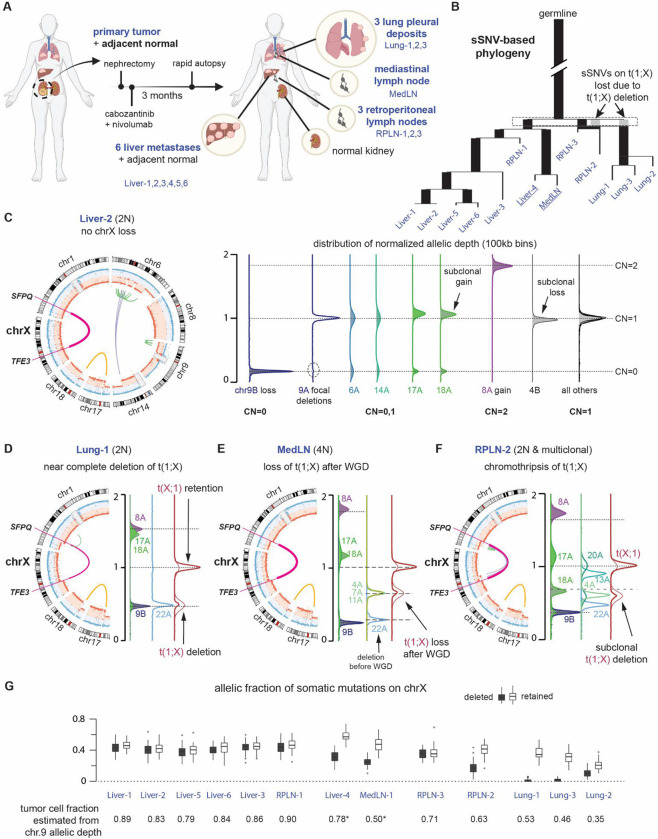
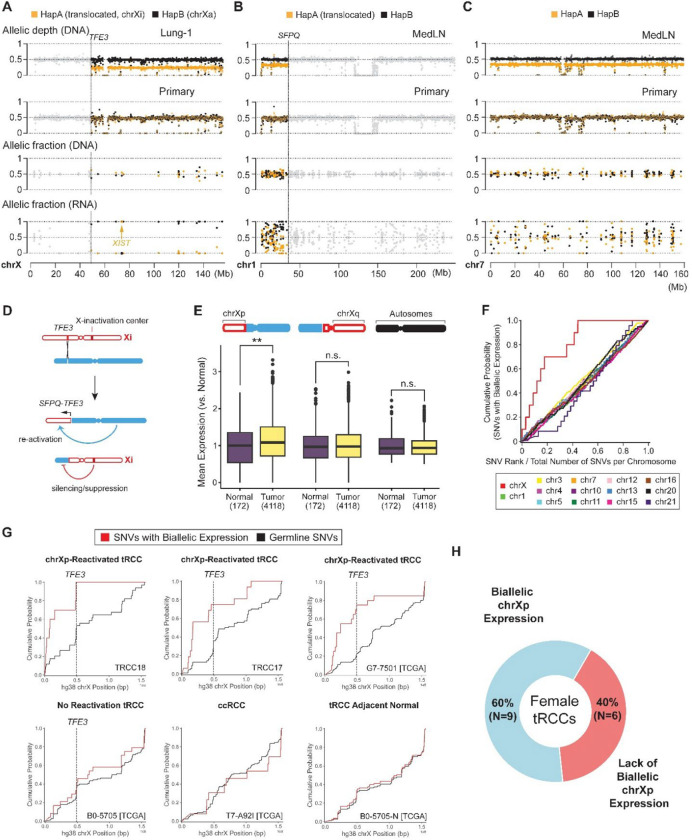
Xp11 translocation renal cell carcinoma (tRCC) is a female-predominant kidney cancer driven by translocations between the TFE3 gene on chromosome Xp11.2 and partner genes located on either chrX or on autosomes. The rearrangement processes that underlie TFE3 fusions, and whether they are linked to the female sex bias of this cancer, are largely unexplored. Moreover, whether oncogenic TFE3 fusions arise from both the active and inactive X chromosomes in females remains unknown. Here we address these questions by haplotype-specific analyses of whole-genome sequences of 29 tRCC samples from 15 patients and by re-analysis of 145 published tRCC whole-exome sequences. We show that TFE3 fusions universally arise as reciprocal translocations with minimal DNA loss or insertion at paired break ends. Strikingly, we observe a near exact 2:1 female:male ratio in TFE3 fusions arising via X:autosomal translocation (but not via X inversion), which accounts for the female predominance of tRCC. This 2:1 ratio is at least partially attributable to oncogenic fusions involving the inactive X chromosome and is accompanied by partial re-activation of silenced chrX genes on the rearranged chromosome. Our results highlight how somatic alterations involving the X chromosome place unique constraints on tumor initiation and exemplify how genetic rearrangements of the sex chromosomes can underlie cancer sex differences.
Conflict of interest statement
Competing Interests: S.R.V. has consulted for Jnana Therapeutics, MPM Capital, and Vida Ventures within the past 3 years; receives research support from Bayer; and his spouse is an employee of and holds equity in Kojin Therapeutics. C.-Z. Zhang is a co-founder, consultant, and equity holder of Pillar Biosciences, a for profit company specialized in assay development for targeted DNA sequencing. T.K.C.: Institutional and/or personal, paid and/or unpaid support for research, advisory boards, consultancy, and/or honoraria past 5 years and ongoing, from: Alkermes, AstraZeneca, Aravive, Aveo, Bayer, Bristol Myers-Squibb, Calithera, Circle Pharma, Deciphera Pharmaceuticals, Eisai, EMD Serono, Exelixis, GlaxoSmithKline, Gilead, IQVA, Infinity, Ipsen, Jansen, Kanaph, Lilly, Merck, Nikang, Nuscan, Novartis, Oncohost, Pfizer, Roche, Sanofi/Aventis, Scholar Rock, Surface Oncology, Takeda, Tempest, Up-To-Date, CME events (Peerview, OncLive, MJH, CCO and others), outside the submitted work. Institutional patents filed on molecular alterations and immunotherapy response/toxicity, and ctDNA. Equity: Tempest, Pionyr, Osel, Precede Bio, CureResponse, InnDura. Committees: NCCN, GU Steering Committee, ASCO/ESMO, ACCRU, KidneyCan. • Medical writing and editorial assistance support may have been funded by Communications companies in part. No speaker’s bureau. Mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/Foreign Components. The institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies or/and royalties potentially involved in research around the subject matter. D.J.E.: Research funding to institution from Bristol-Myers Squibb, Cardiff Oncology, MiNK Therapeutics, Novartis, Sanofi, Puma. Discounted research sequencing from Foundation Medicine.
Figures
References
-
- Argani P., MiT family translocation renal cell carcinoma. Seminars in Diagnostic Pathology. 32, 103–113 (2015). - PubMed
-
- Choueiri T. K., Lim Z. D., Hirsch M. S., Tamboli P., Jonasch E., McDermott D. F., Dal Cin P., Corn P., Vaishampayan U., Heng D. Y. C., Tannir N. M., Vascular endothelial growth factor-targeted therapy for the treatment of adult metastatic Xp11.2 translocation renal cell carcinoma. Cancer. 116, 5219–5225 (2010). - PMC - PubMed
-
- Malouf G. G., Camparo P., Oudard S., Schleiermacher G., Theodore C., Rustine A., Dutcher J., Billemont B., Rixe O., Bompas E., Guillot A., Boccon-Gibod L., Couturier J., Molinié V., Escudier B., Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol. 21, 1834–1838 (2010). - PubMed
-
- Boilève A., Carlo M. I., Barthélémy P., Oudard S., Borchiellini D., Voss M. H., George S., Chevreau C., Landman-Parker J., Tabone M.-D., Chism D. D., Amin A., Bilen M. A., Bosse D., Coulomb-L’hermine A., Su X., Choueiri T. K., Tannir N. M., Malouf G. G., Immune checkpoint inhibitors in MITF family translocation renal cell carcinomas and genetic correlates of exceptional responders. j. immunotherapy cancer. 6, 159 (2018). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources