

This is a preprint.
Conserved regulatory motifs in the juxtamembrane domain and kinase N-lobe revealed through deep mutational scanning of the MET receptor tyrosine kinase domain
- PMID: 37577651
- PMCID: PMC10418267
- DOI: 10.1101/2023.08.03.551866
Conserved regulatory motifs in the juxtamembrane domain and kinase N-lobe revealed through deep mutational scanning of the MET receptor tyrosine kinase domain
Update in
-
Conserved regulatory motifs in the juxtamembrane domain and kinase N-lobe revealed through deep mutational scanning of the MET receptor tyrosine kinase domain.Elife. 2024 Sep 13;12:RP91619. doi: 10.7554/eLife.91619. Elife. 2024. PMID: 39268701 Free PMC article.
Abstract
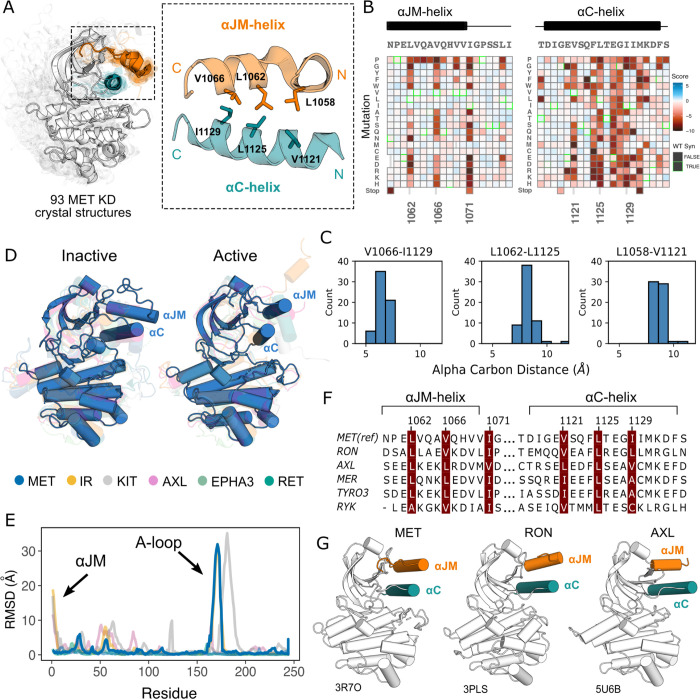
MET is a receptor tyrosine kinase (RTK) responsible for initiating signaling pathways involved in development and wound repair. MET activation relies on ligand binding to the extracellular receptor, which prompts dimerization, intracellular phosphorylation, and recruitment of associated signaling proteins. Mutations, which are predominantly observed clinically in the intracellular juxtamembrane and kinase domains, can disrupt typical MET regulatory mechanisms. Understanding how juxtamembrane variants, such as exon 14 skipping (METΔEx14), and rare kinase domain mutations can increase signaling, often leading to cancer, remains a challenge. Here, we perform a parallel deep mutational scan (DMS) of the MET intracellular kinase domain in two fusion protein backgrounds: wild type and METΔEx14. Our comparative approach has revealed a critical hydrophobic interaction between a juxtamembrane segment and the kinase αC-helix, pointing to potential differences in regulatory mechanisms between MET and other RTKs. Additionally, we have uncovered a β5 motif that acts as a structural pivot for the kinase domain in MET and other TAM family of kinases. We also describe a number of previously unknown activating mutations, aiding the effort to annotate driver, passenger, and drug resistance mutations in the MET kinase domain.
Conflict of interest statement
Competing Interests JSF is a consultant for, has equity in, and receives research support from Relay Therapeutics. N.J. is a founder of Rezo Therapeutics and a shareholder of Rezo Therapeutics, Sudo Therapeutics, and Type6 Therapeutics. N.J. is a SAB member of Sudo Therapeutics, Type6 Therapeutic and NIBR Oncology. The Jura laboratory has received sponsored research support from Genentech, Rezo Therapeutics and Type6 Therapeutics. E.A.C. is a consultant at IHP Therapeutics, Valar Labs, Tatara Therapeutics and Pear Diagnostics, reports receiving commercial research grants from Pfizer, and has stock ownership in Tatara Therapeutics, HDT Bio, Clara Health, Aqtual, and Guardant Health.
Figures





References
-
- Abella J. V., Peschard P., Naujokas M. A., Lin T., Saucier C., Urbé S., & Park M. (2005). Met/Hepatocyte Growth Factor Receptor Ubiquitination Suppresses Transformation and Is Required for Hrs Phosphorylation. Molecular and Cellular Biology, 25(21), 9632–9645. 10.1128/MCB.25.21.9632-9645.2005 - DOI - PMC - PubMed
-
- Ahler E., Register A. C., Chakraborty S., Fang L., Dieter E. M., Sitko K. A., Vidadala R. S. R., Trevillian B. M., Golkowski M., Gelman H., Stephany J. J., Rubin A. F., Merritt E. A., Fowler D. M., & Maly D. J. (2019). A Combined Approach Reveals a Regulatory Mechanism Coupling Src’s Kinase Activity, Localization, and Phosphotransferase-Independent Functions. Molecular Cell, 74(2), 393–408.e20. 10.1016/j.molcel.2019.02.003 - DOI - PMC - PubMed
-
- Andrews S., & Others. (2010). “FastQC: A Quality Control Tool for High Throughput Sequence Data.” Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Bardelli A., Longati P., Gramaglia D., Basilico C., Tamagnone L., Giordano S., Ballinari D., Michieli P., & Comoglio P. M. (1998). Uncoupling signal transducers from oncogenic MET mutants abrogates cell transformation and inhibits invasive growth. Proceedings of the National Academy of Sciences, 95(24), 14379–14383. 10.1073/pnas.95.24.14379 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous