

Nuclear proinflammatory cytokine S100A9 enhances expression of human papillomavirus oncogenes via transcription factor TEAD1
- PMID: 37578237
- PMCID: PMC10506480
- DOI: 10.1128/jvi.00815-23
Nuclear proinflammatory cytokine S100A9 enhances expression of human papillomavirus oncogenes via transcription factor TEAD1
Erratum in
-
Correction for Mori et al., "Nuclear proinflammatory cytokine S100A9 enhances expression of human papillomavirus oncogenes via transcription factor TEAD1".J Virol. 2023 Nov 30;97(11):e0149923. doi: 10.1128/jvi.01499-23. Epub 2023 Oct 26. J Virol. 2023. PMID: 37882517 Free PMC article. No abstract available.
Abstract
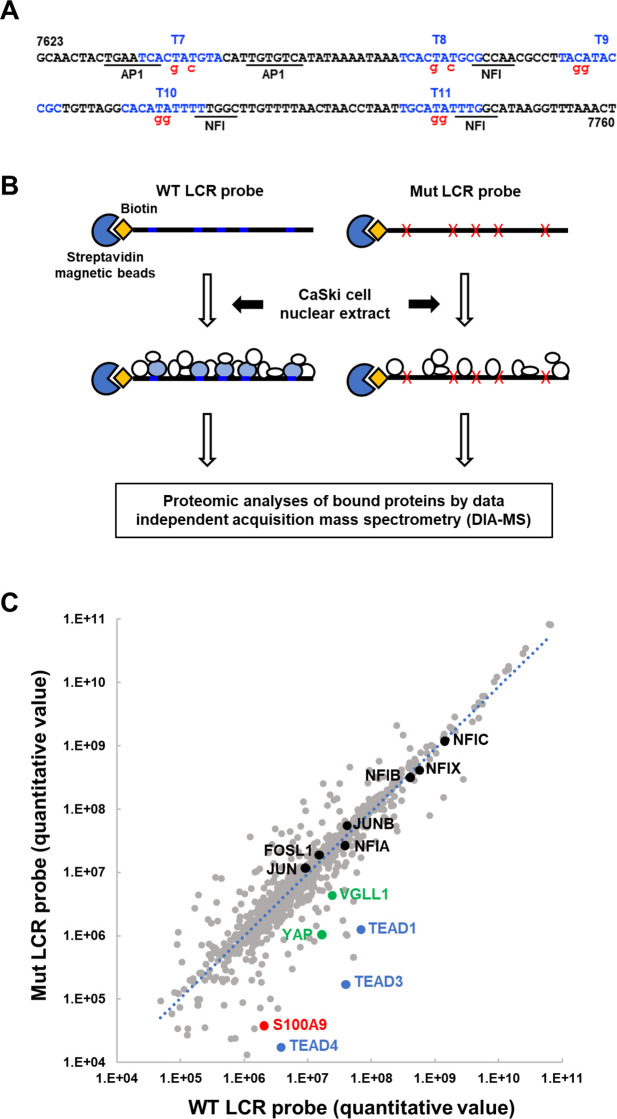
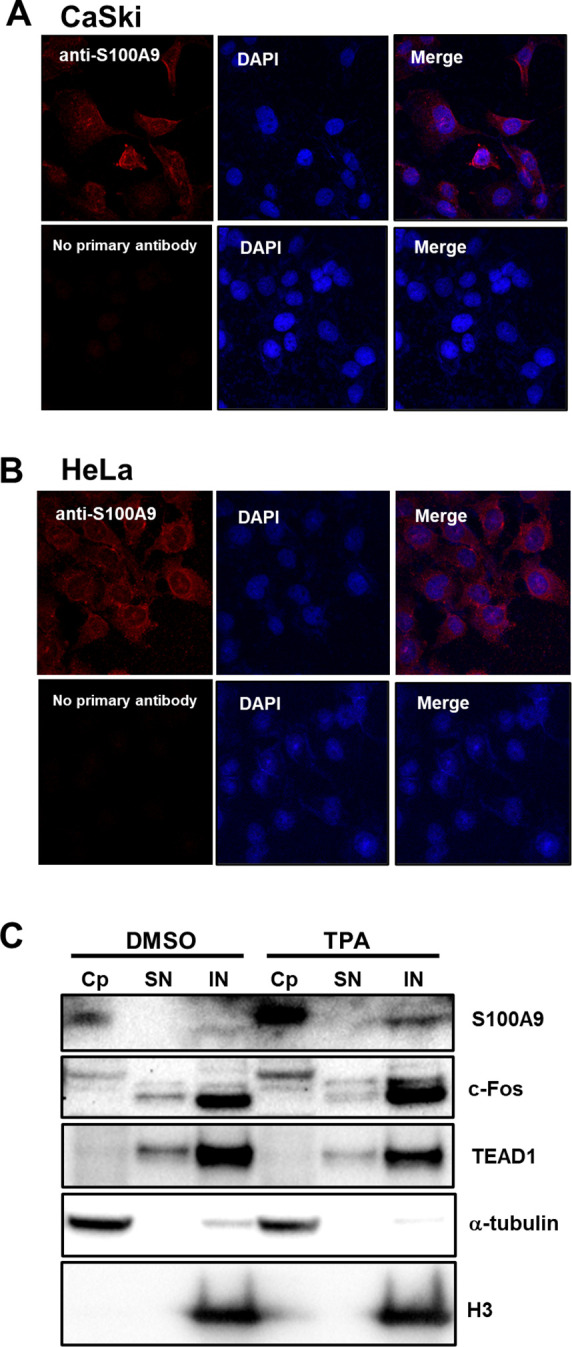
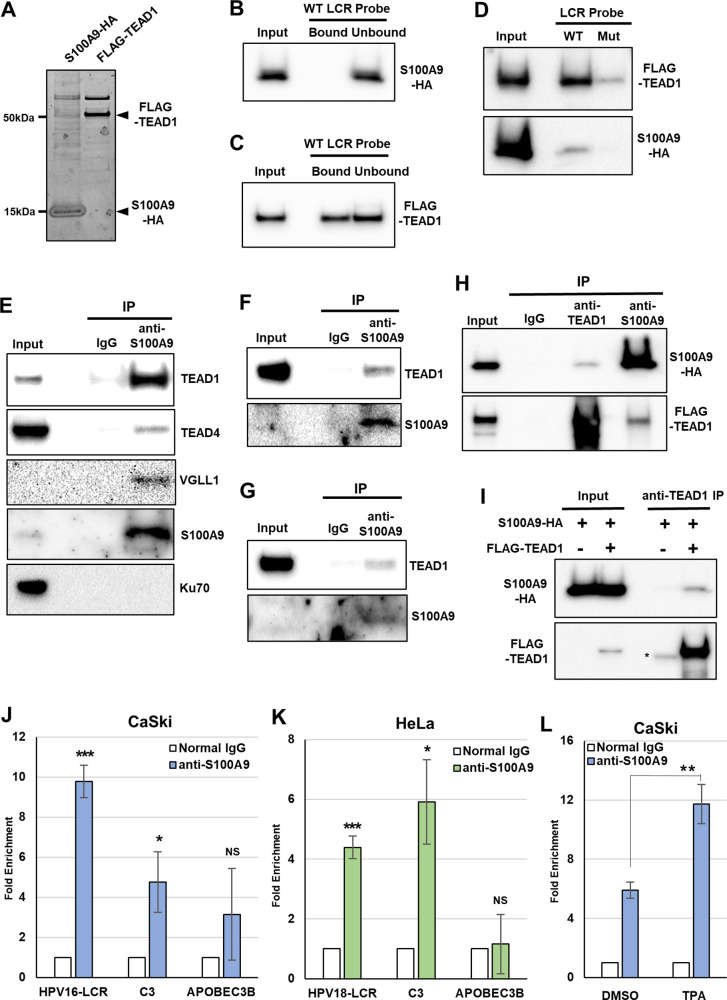
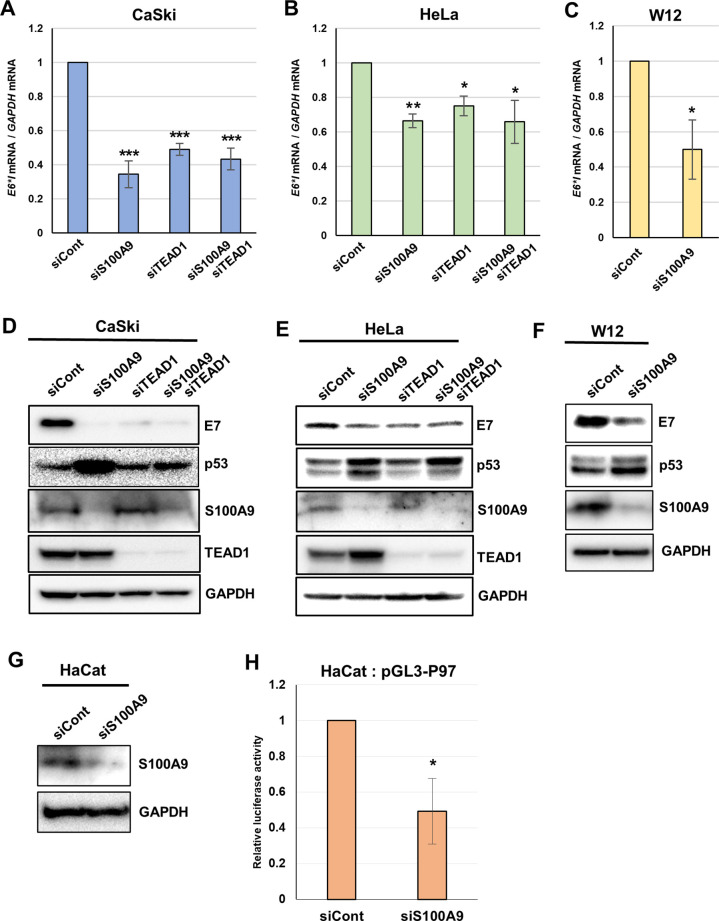
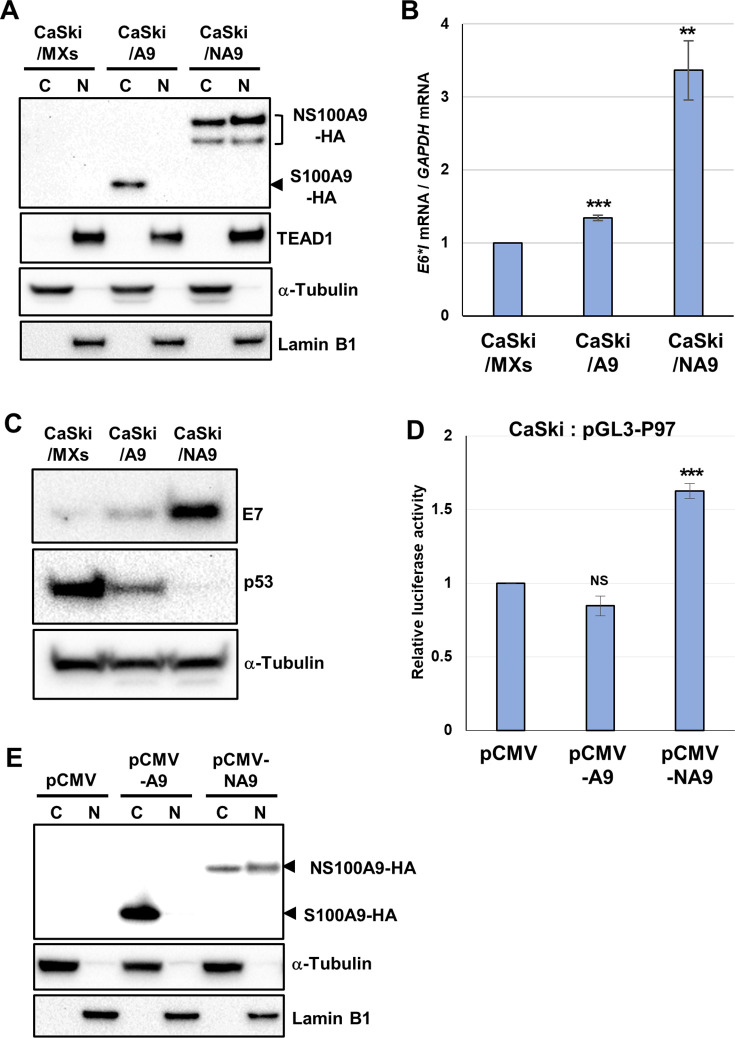
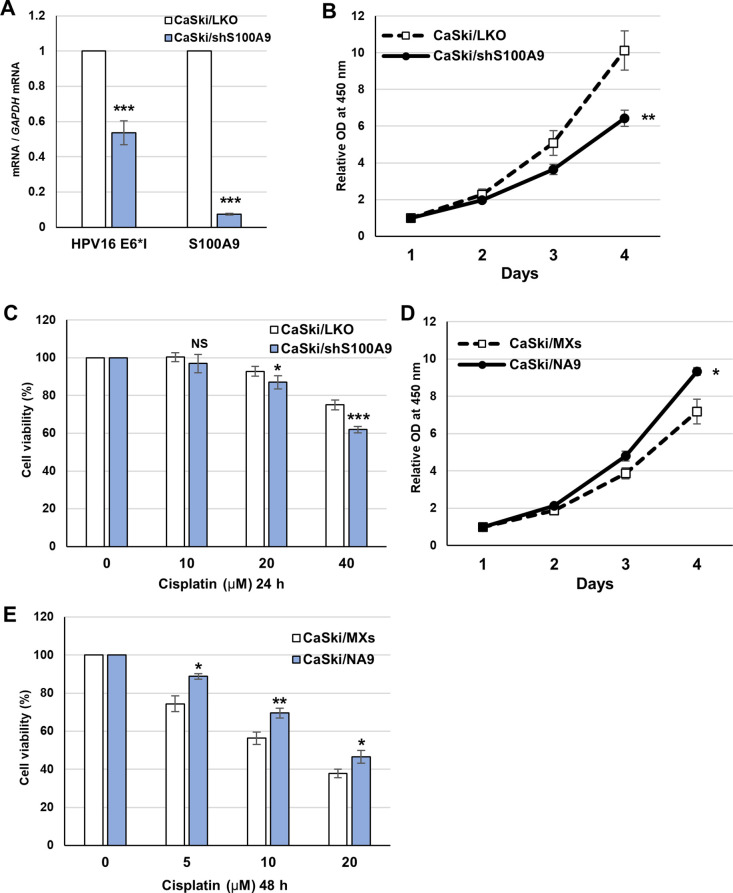
Transcription of the human papillomavirus (HPV) oncogenes, E6 and E7, is regulated by the long control region (LCR) of the viral genome. Although various transcription factors have been reported to bind to the LCR, little is known about the transcriptional cofactors that modulate HPV oncogene expression in association with these transcription factors. Here, we performed in vitro DNA-pulldown purification of nuclear proteins in cervical cancer cells, followed by proteomic analyses to identify transcriptional cofactors that bind to the HPV16 LCR via the transcription factor TEAD1. We detected the proinflammatory cytokine S100A9 that localized to the nucleus of cervical cancer cells and associated with the LCR via direct interaction with TEAD1. Nuclear S100A9 levels and its association with the LCR were increased in cervical cancer cells by treatment with a proinflammatory phorbol ester. Knockdown of S100A9 decreased HPV oncogene expression and reduced the growth of cervical cancer cells and their susceptibility to cisplatin, whereas forced nuclear expression of S100A9 using nuclear localization signals exerted opposite effects. Thus, we conclude that nuclear S100A9 binds to the HPV LCR via TEAD1 and enhances viral oncogene expression by acting as a transcriptional coactivator. IMPORTANCE Human papillomavirus (HPV) infection is the primary cause of cervical cancer, and the viral oncogenes E6 and E7 play crucial roles in carcinogenesis. Although cervical inflammation contributes to the development of cervical cancer, the molecular mechanisms underlying the role of these inflammatory responses in HPV carcinogenesis are not fully understood. Our study shows that S100A9, a proinflammatory cytokine, is induced in the nucleus of cervical cancer cells by inflammatory stimuli, and it enhances HPV oncogene expression by acting as a transcriptional coactivator of TEAD1. These findings provide new molecular insights into the relationship between inflammation and viral carcinogenesis.
Keywords: HPV; S100A9; TEAD1; proinflammatory cytokine; transcriptional coactivator; viral oncogenes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The Transcriptional Cofactor VGLL1 Drives Transcription of Human Papillomavirus Early Genes via TEAD1.J Virol. 2020 May 4;94(10):e01945-19. doi: 10.1128/JVI.01945-19. Print 2020 May 4. J Virol. 2020. PMID: 32132238 Free PMC article.
-
Nanog, in Cooperation with AP1, Increases the Expression of E6/E7 Oncogenes from HPV Types 16/18.Viruses. 2021 Jul 28;13(8):1482. doi: 10.3390/v13081482. Viruses. 2021. PMID: 34452350 Free PMC article.
-
The genetic variability, phylogeny and functional significance of E6, E7 and LCR in human papillomavirus type 52 isolates in Sichuan, China.Virol J. 2021 May 3;18(1):94. doi: 10.1186/s12985-021-01565-5. Virol J. 2021. PMID: 33941222 Free PMC article.
-
Protein-DNA Interactions Regulate Human Papillomavirus DNA Replication, Transcription, and Oncogenesis.Int J Mol Sci. 2023 May 9;24(10):8493. doi: 10.3390/ijms24108493. Int J Mol Sci. 2023. PMID: 37239839 Free PMC article. Review.
-
Molecular mechanism of carcinogenesis by human papillomavirus-16.J Dermatol. 2000 Feb;27(2):73-86. doi: 10.1111/j.1346-8138.2000.tb02126.x. J Dermatol. 2000. PMID: 10721654 Review.
Cited by
-
Deciphering the divergent transcriptomic landscapes of cervical cancer cells grown in 3D and 2D cell culture systems.Front Cell Dev Biol. 2024 Aug 13;12:1413882. doi: 10.3389/fcell.2024.1413882. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39193365 Free PMC article.
-
Manipulating host secreted protein gene expression: an indirect approach by HPV11/16 E6/E7 to suppress PBMC cytokine secretion.Virol J. 2024 Aug 2;21(1):172. doi: 10.1186/s12985-024-02432-9. Virol J. 2024. PMID: 39095779 Free PMC article.
-
Insights into expression and localization of HPV16 LCR-associated transcription factors and association with LCR activity in HNSCC.Mol Ther Oncol. 2024 Dec 21;33(1):200926. doi: 10.1016/j.omton.2024.200926. eCollection 2025 Mar 20. Mol Ther Oncol. 2024. PMID: 39886356 Free PMC article.
-
Nuclear keratin 6A upregulates human papillomavirus oncogene expression through TEAD1 interaction.Virol J. 2025 Jun 21;22(1):202. doi: 10.1186/s12985-025-02832-5. Virol J. 2025. PMID: 40544293 Free PMC article.
-
Deciphering the tumor immune microenvironment: single-cell and spatial transcriptomic insights into cervical cancer fibroblasts.J Exp Clin Cancer Res. 2025 Jul 5;44(1):194. doi: 10.1186/s13046-025-03432-5. J Exp Clin Cancer Res. 2025. PMID: 40616092 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
