

Simultaneous inhibition of DNA-PK and Polϴ improves integration efficiency and precision of genome editing
- PMID: 37580318
- PMCID: PMC10425386
- DOI: 10.1038/s41467-023-40344-4
Simultaneous inhibition of DNA-PK and Polϴ improves integration efficiency and precision of genome editing
Abstract
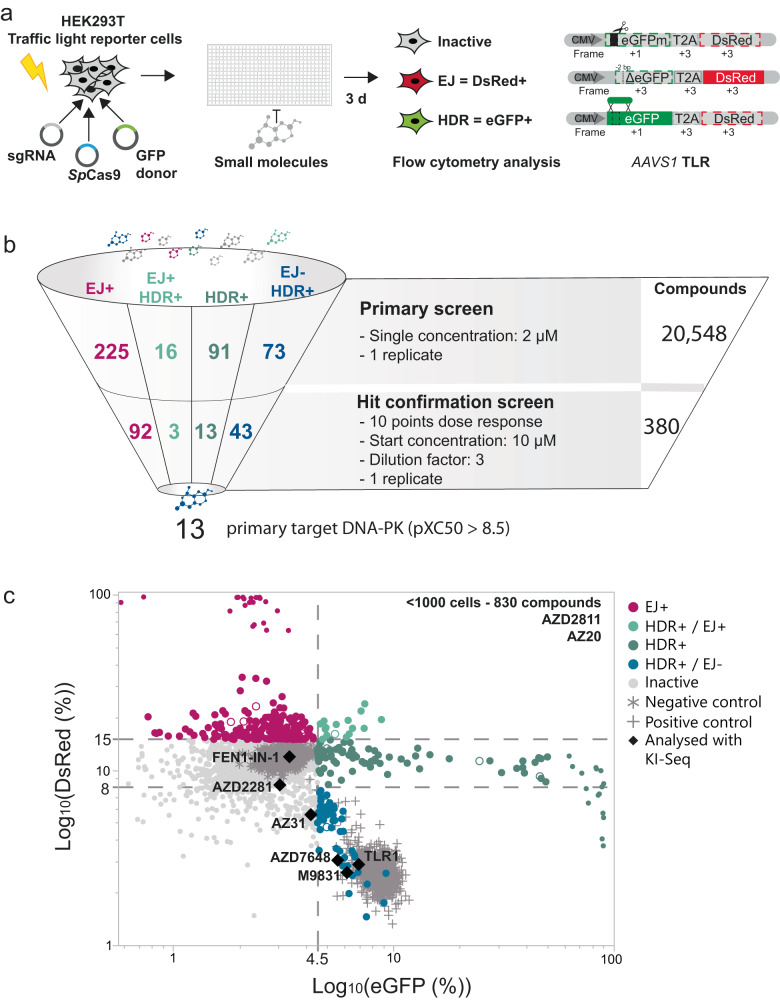
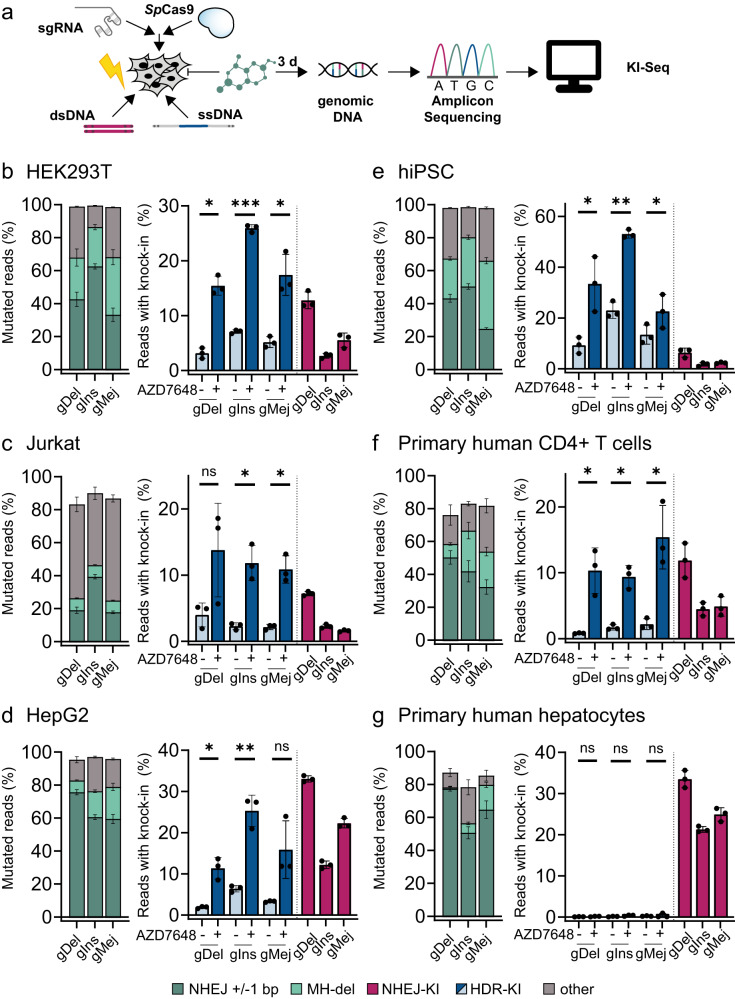
Genome editing, specifically CRISPR/Cas9 technology, has revolutionized biomedical research and offers potential cures for genetic diseases. Despite rapid progress, low efficiency of targeted DNA integration and generation of unintended mutations represent major limitations for genome editing applications caused by the interplay with DNA double-strand break repair pathways. To address this, we conduct a large-scale compound library screen to identify targets for enhancing targeted genome insertions. Our study reveals DNA-dependent protein kinase (DNA-PK) as the most effective target to improve CRISPR/Cas9-mediated insertions, confirming previous findings. We extensively characterize AZD7648, a selective DNA-PK inhibitor, and find it to significantly enhance precise gene editing. We further improve integration efficiency and precision by inhibiting DNA polymerase theta (Polϴ). The combined treatment, named 2iHDR, boosts templated insertions to 80% efficiency with minimal unintended insertions and deletions. Notably, 2iHDR also reduces off-target effects of Cas9, greatly enhancing the fidelity and performance of CRISPR/Cas9 gene editing.
© 2023. Springer Nature Limited.
Conflict of interest statement
S.W., N.A., M.F., J.B., S.E., A.L., P.H., S.L., S.C., J.S., B.B., B.S., B.M., S.DC., P.I., M.B., T.M., S.R., O.E., E.B.C., J.V.F., S.Š., P.A., A.T.G. and M.M. are presently or were previously employed by AstraZeneca and may be AstraZeneca shareholders. M.K.S., M.R.S. and TM are presently employed by Promega Corporation. S.W., N.A., S.Š. and M.M. are listed as co-inventors in an AstraZeneca patent (WO2023052508A2) related to this work.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
