

In vivo screening characterizes chromatin factor functions during normal and malignant hematopoiesis
- PMID: 37580596
- PMCID: PMC10484791
- DOI: 10.1038/s41588-023-01471-2
In vivo screening characterizes chromatin factor functions during normal and malignant hematopoiesis
Abstract
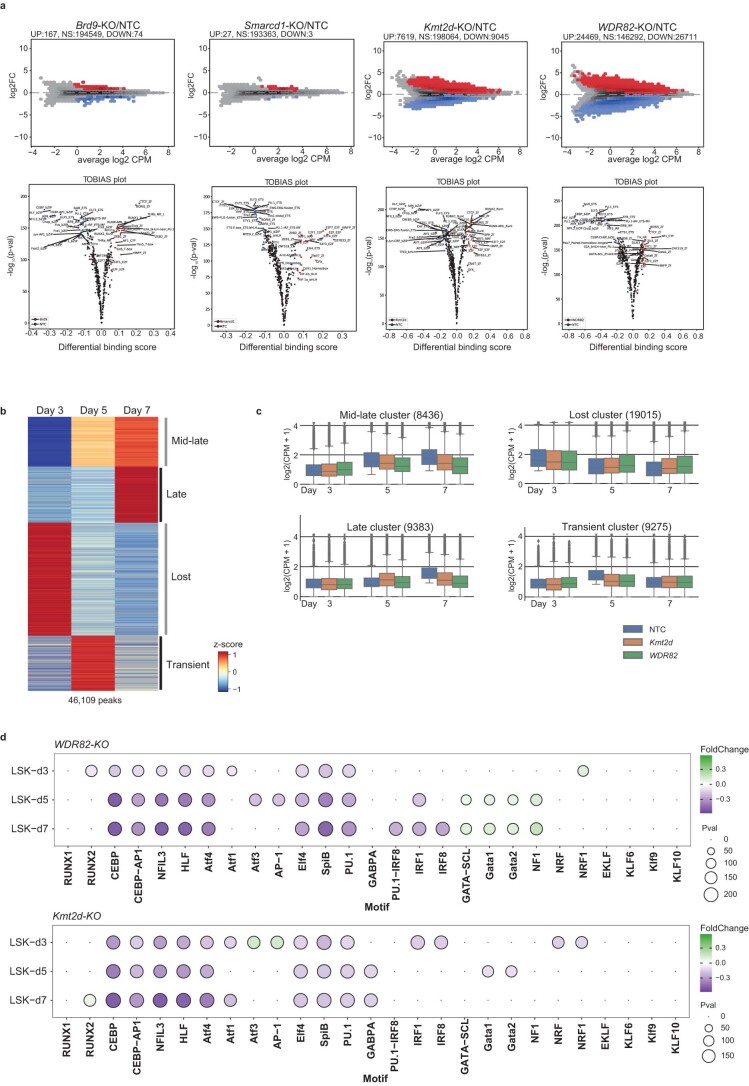
Cellular differentiation requires extensive alterations in chromatin structure and function, which is elicited by the coordinated action of chromatin and transcription factors. By contrast with transcription factors, the roles of chromatin factors in differentiation have not been systematically characterized. Here, we combine bulk ex vivo and single-cell in vivo CRISPR screens to characterize the role of chromatin factor families in hematopoiesis. We uncover marked lineage specificities for 142 chromatin factors, revealing functional diversity among related chromatin factors (i.e. barrier-to-autointegration factor subcomplexes) as well as shared roles for unrelated repressive complexes that restrain excessive myeloid differentiation. Using epigenetic profiling, we identify functional interactions between lineage-determining transcription factors and several chromatin factors that explain their lineage dependencies. Studying chromatin factor functions in leukemia, we show that leukemia cells engage homeostatic chromatin factor functions to block differentiation, generating specific chromatin factor-transcription factor interactions that might be therapeutically targeted. Together, our work elucidates the lineage-determining properties of chromatin factors across normal and malignant hematopoiesis.
© 2023. The Author(s).
Conflict of interest statement
T.G. and J.P.T.-K. are employees receiving compensation from Relation Therapeutics. J.P.T.-K. is a founder of Relation Therapeutics. D.L.-A. is a consultant of Relation Therapeutics. The other authors declare no competing interests.
Figures

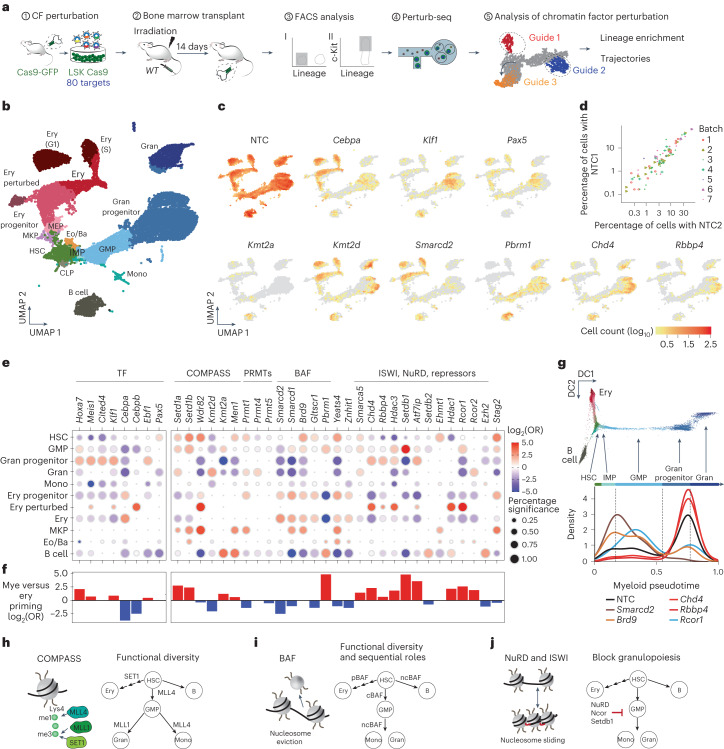
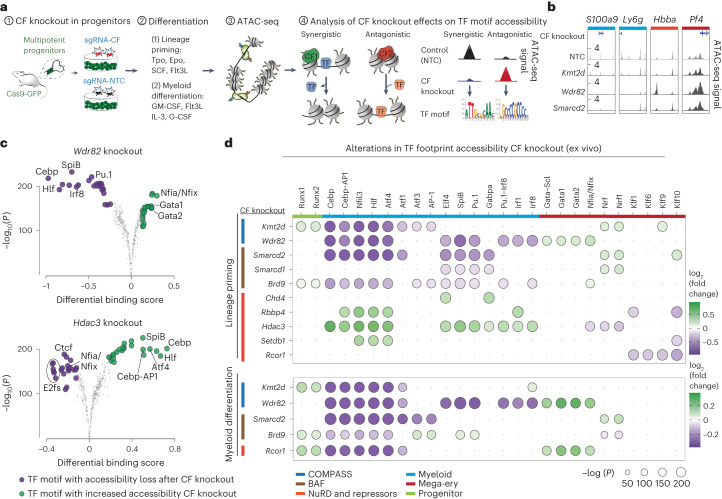


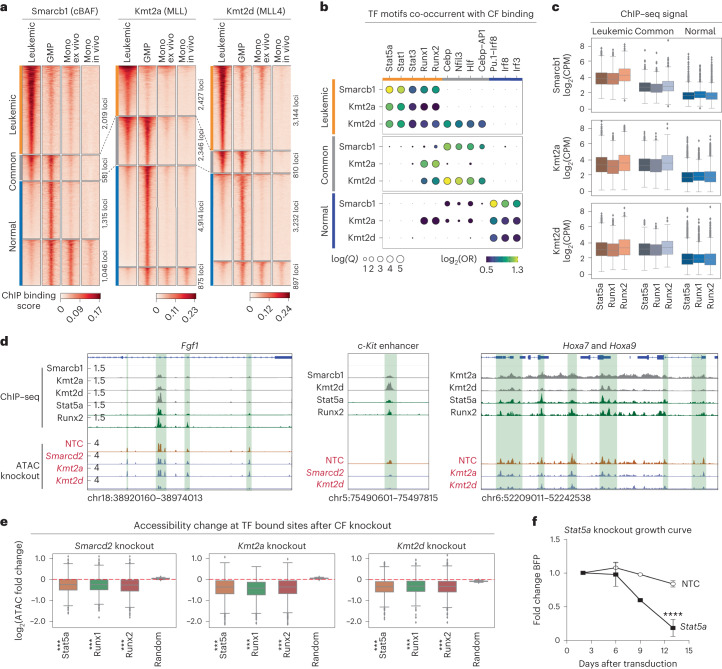
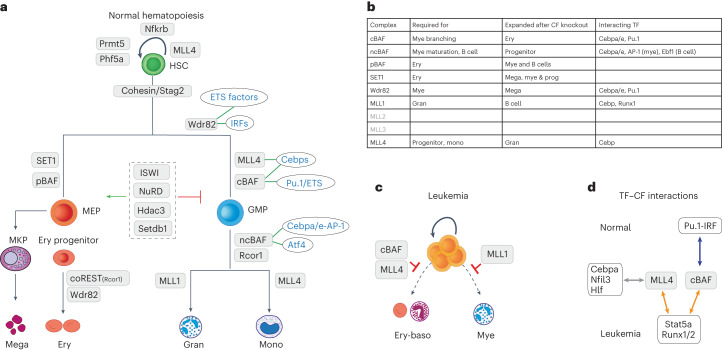









References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
