

SPOP Mutations Target STING1 Signaling in Prostate Cancer and Create Therapeutic Vulnerabilities to PARP Inhibitor-Induced Growth Suppression
- PMID: 37581614
- PMCID: PMC11017857
- DOI: 10.1158/1078-0432.CCR-23-1439
SPOP Mutations Target STING1 Signaling in Prostate Cancer and Create Therapeutic Vulnerabilities to PARP Inhibitor-Induced Growth Suppression
Abstract
Purpose: Speckle-type POZ protein (SPOP) is important in DNA damage response (DDR) and maintenance of genomic stability. Somatic heterozygous missense mutations in the SPOP substrate-binding cleft are found in up to 15% of prostate cancers. While mutations in SPOP predict for benefit from androgen receptor signaling inhibition (ARSi) therapy, outcomes for patients with SPOP-mutant (SPOPmut) prostate cancer are heterogeneous and targeted treatments for SPOPmut castrate-resistant prostate cancer (CRPC) are lacking.
Experimental design: Using in silico genomic and transcriptomic tumor data, proteomics analysis, and genetically modified cell line models, we demonstrate mechanistic links between SPOP mutations, STING signaling alterations, and PARP inhibitor vulnerabilities.
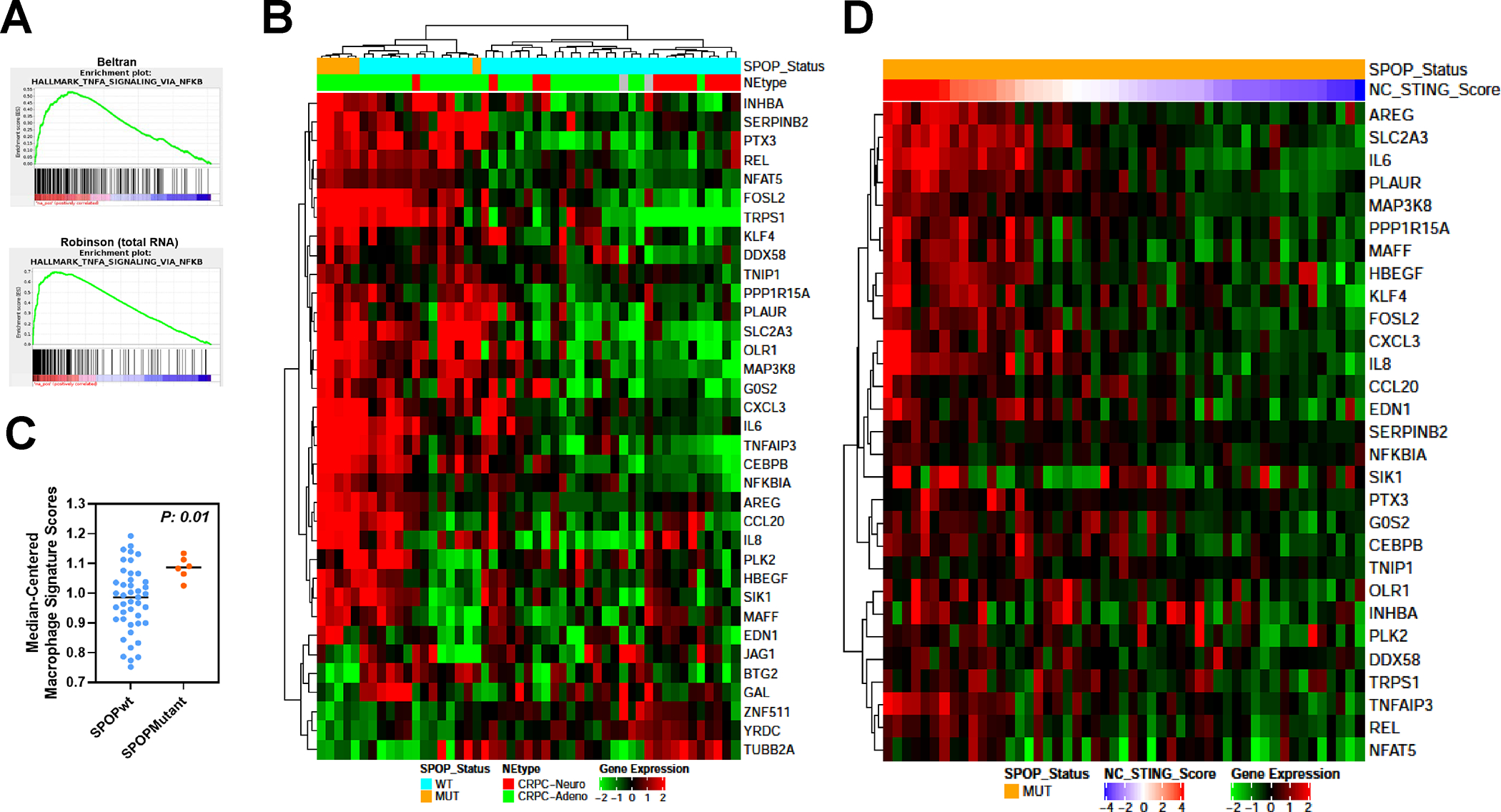
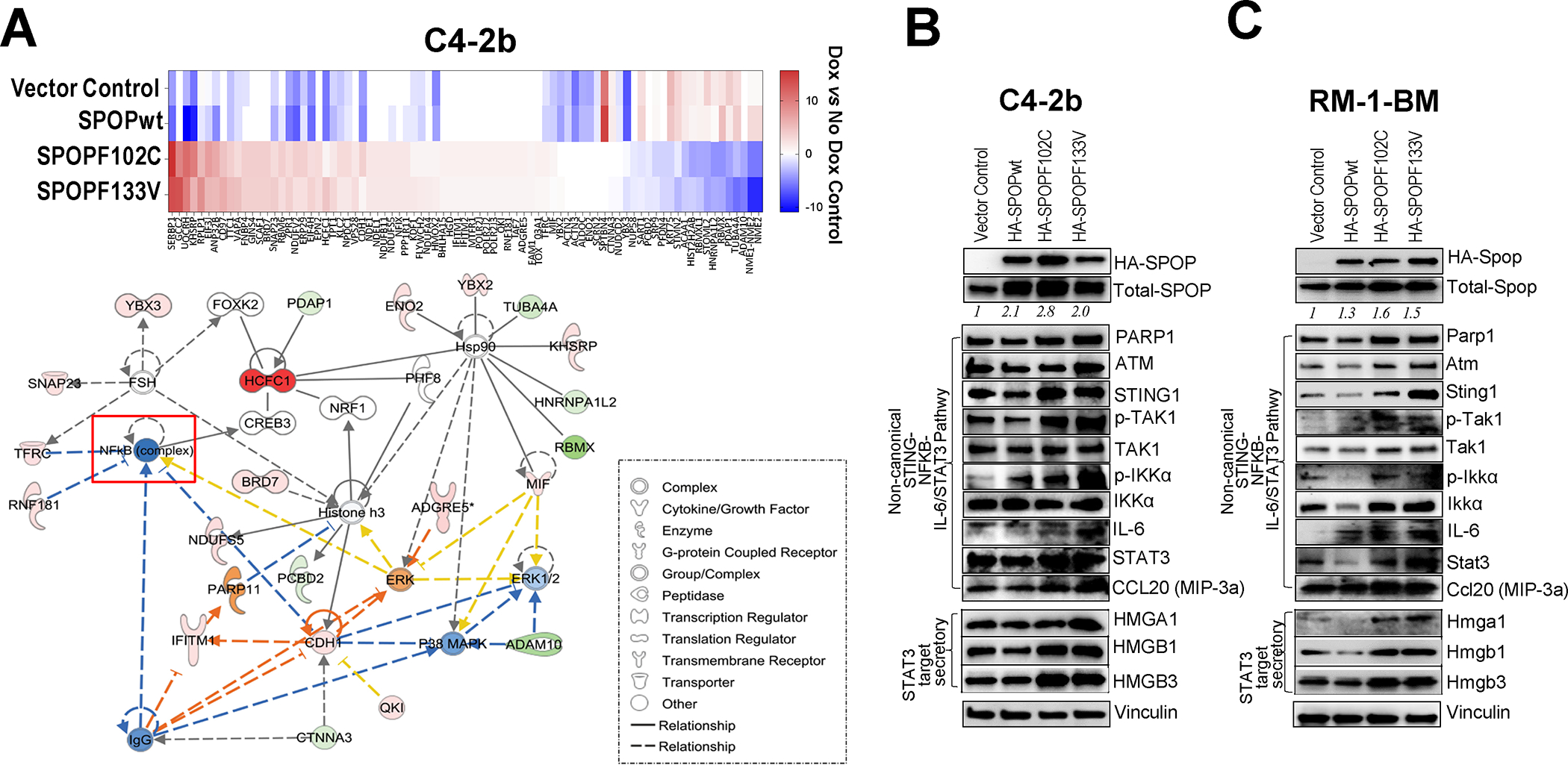
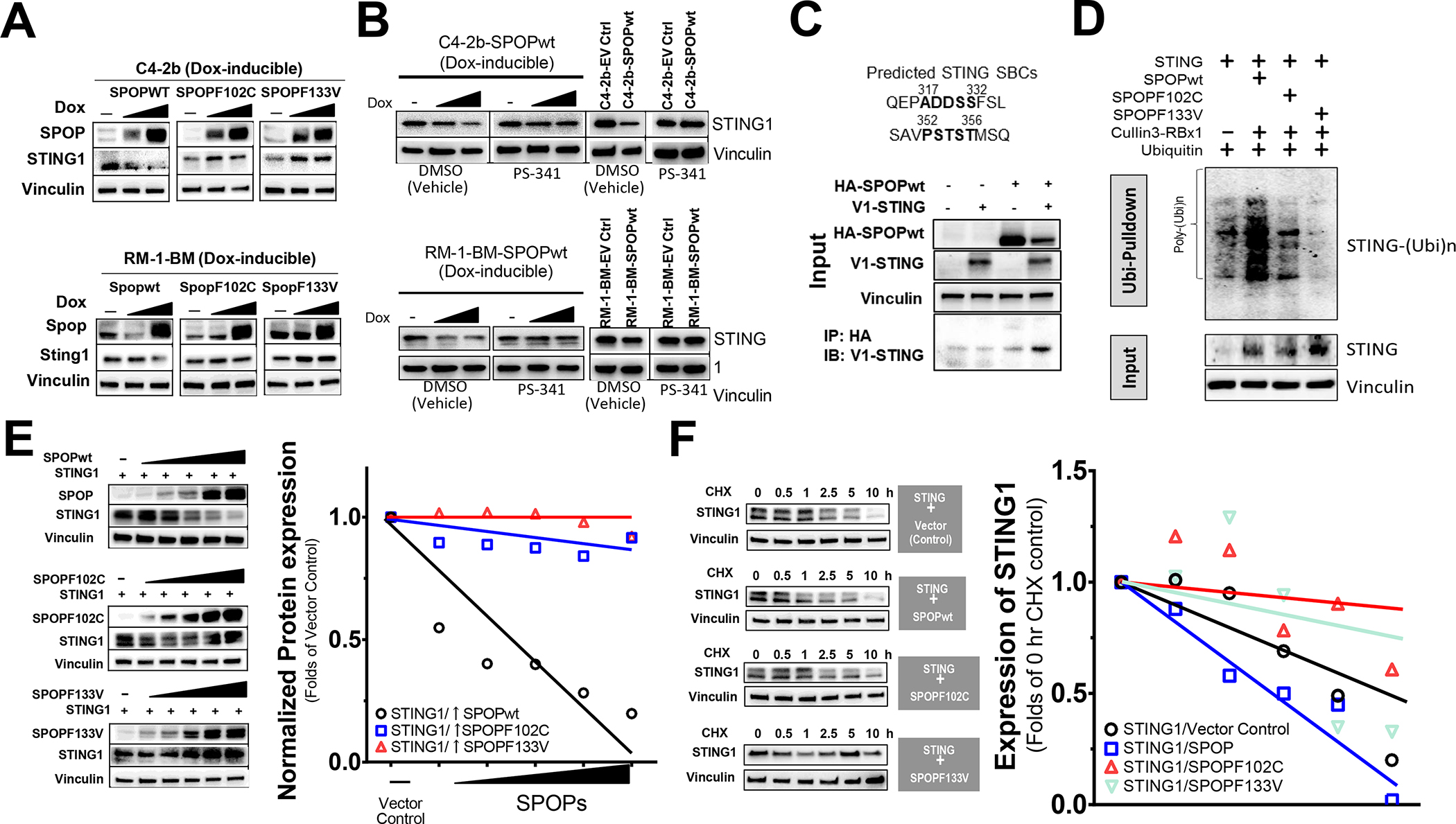
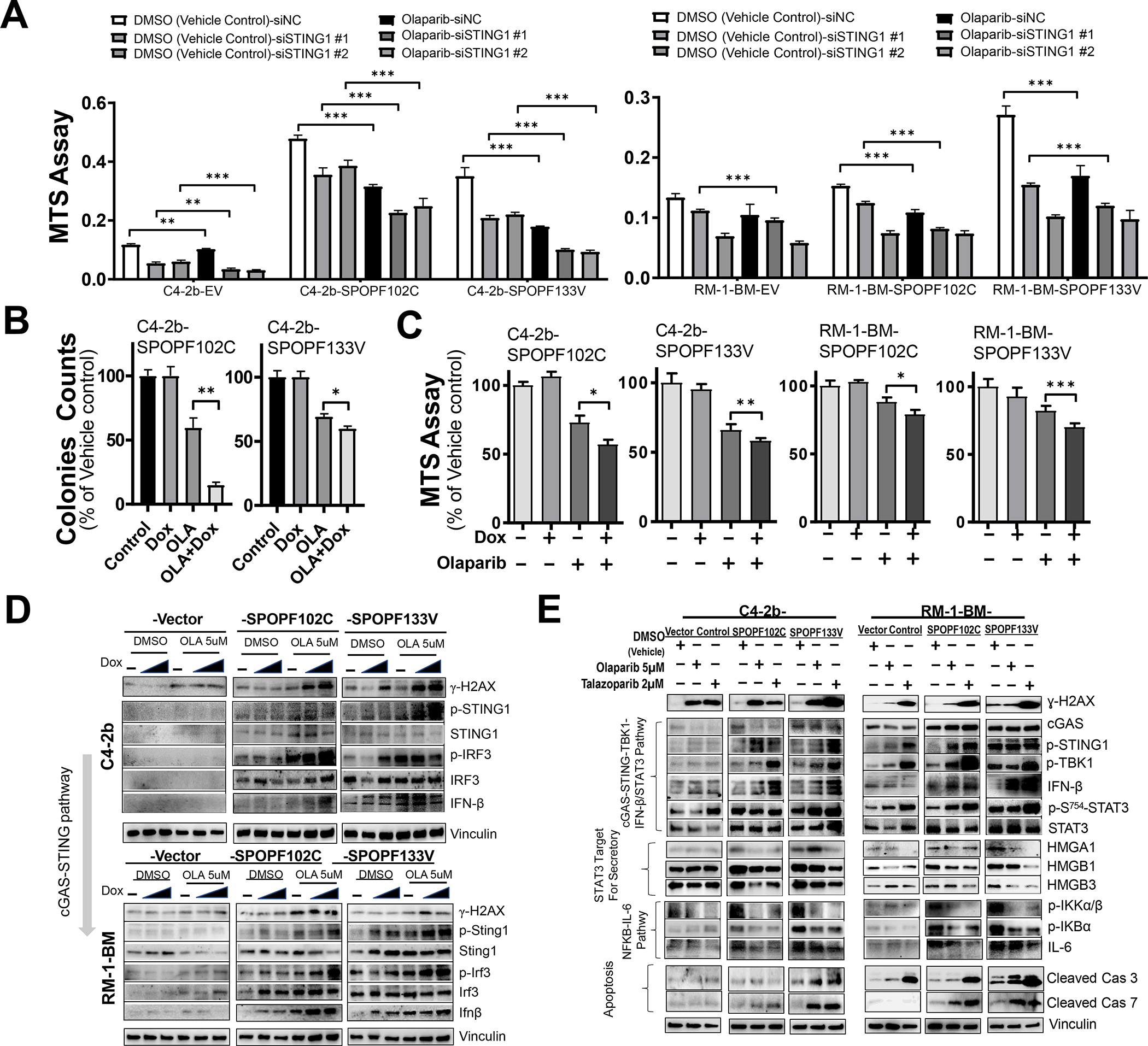
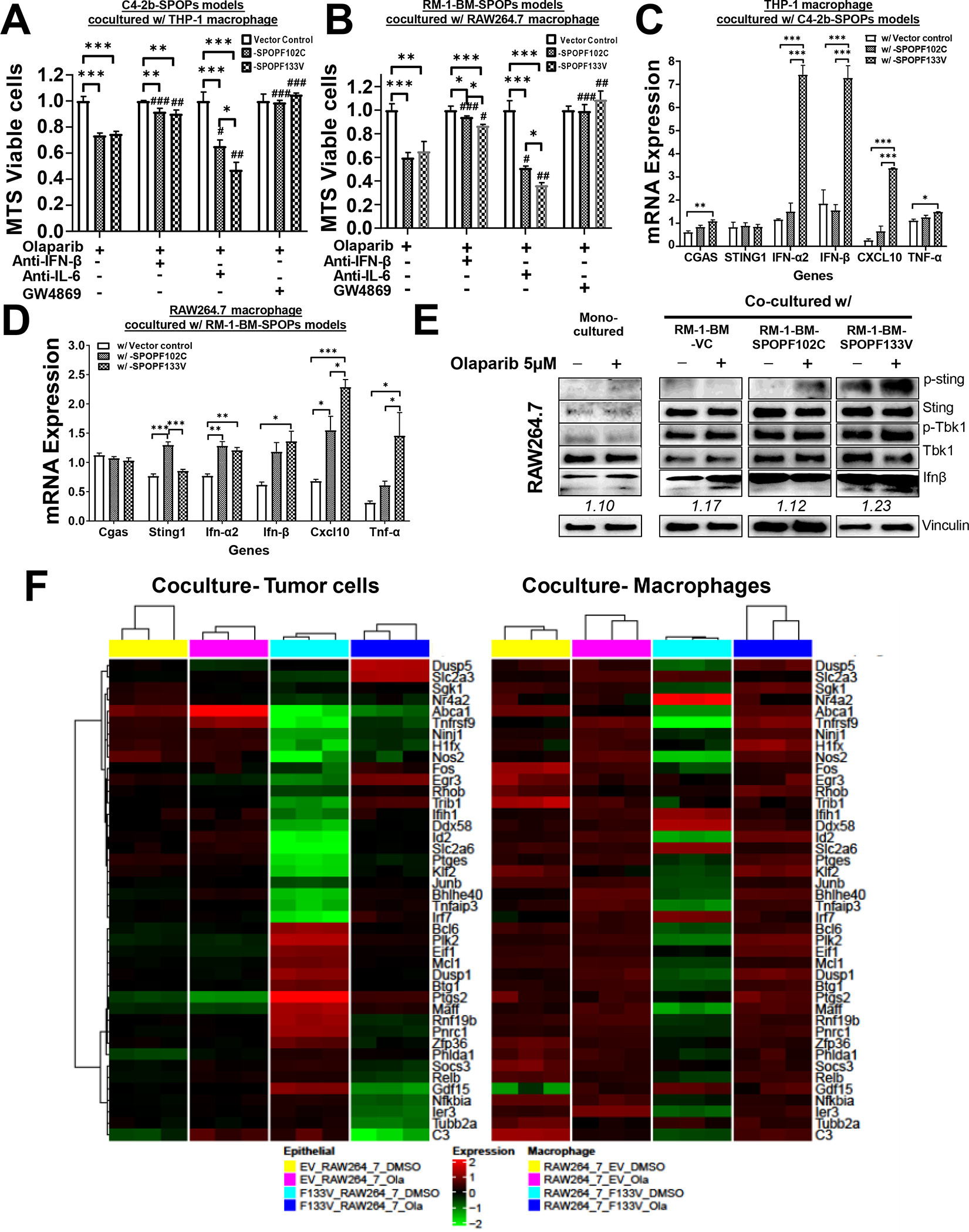
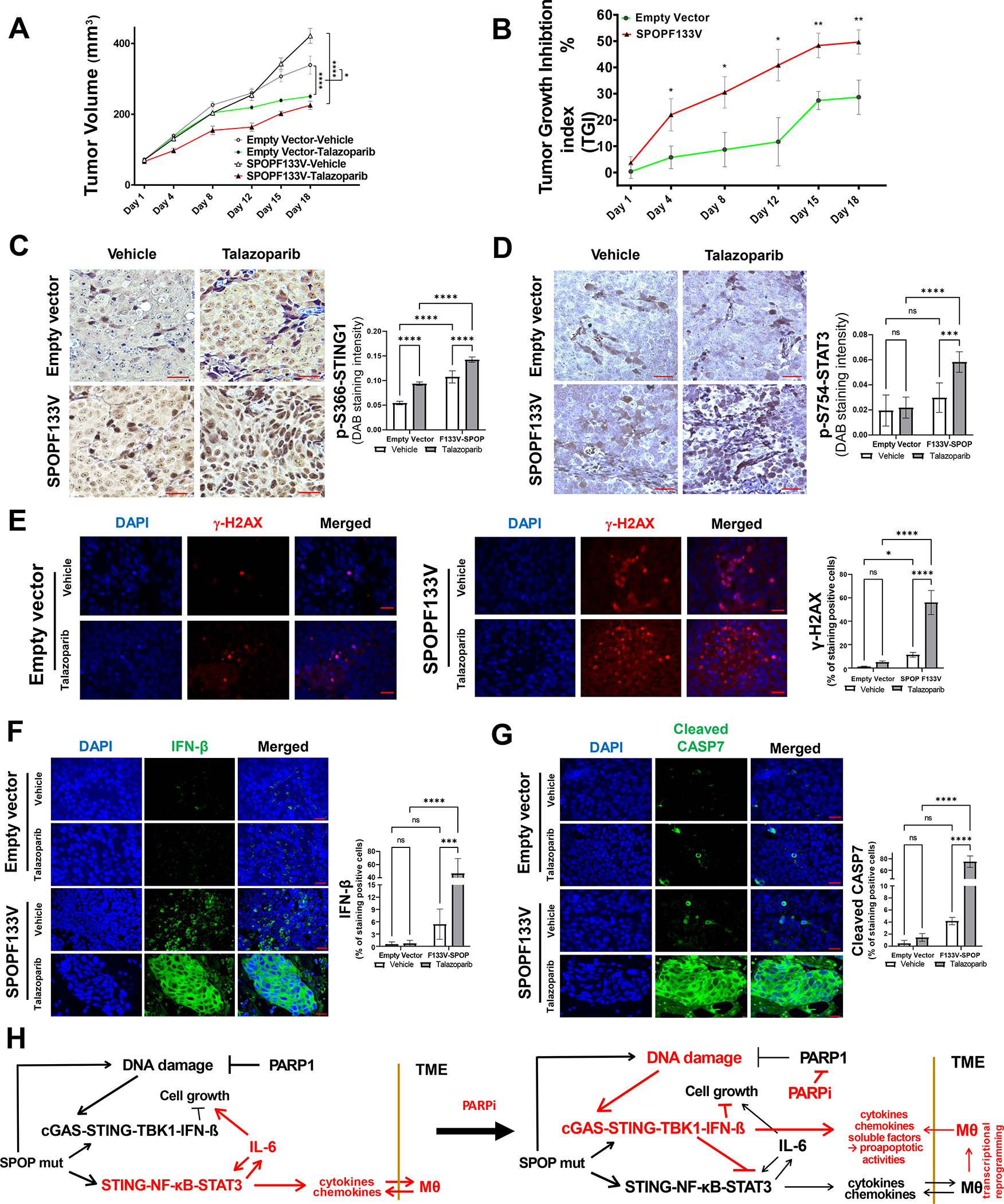
Results: We demonstrate that SPOP mutations are associated with upregulation of a 29-gene noncanonical (NC) STING (NC-STING) signature in a subset of SPOPmut, treatment-refractory CRPC patients. We show in preclinical CRPC models that SPOP targets and destabilizes STING1 protein, and prostate cancer-associated SPOP mutations result in upregulated NC-STING-NF-κB signaling and macrophage- and tumor microenvironment (TME)-facilitated reprogramming, leading to tumor cell growth. Importantly, we provide in vitro and in vivo mechanism-based evidence that PARP inhibitor (PARPi) treatment results in a shift from immunosuppressive NC-STING-NF-κB signaling to antitumor, canonical cGAS-STING-IFNβ signaling in SPOPmut CRPC and results in enhanced tumor growth inhibition.
Conclusions: We provide evidence that SPOP is critical in regulating immunosuppressive versus antitumor activity downstream of DNA damage-induced STING1 activation in prostate cancer. PARPi treatment of SPOPmut CRPC alters this NC-STING signaling toward canonical, antitumor cGAS-STING-IFNβ signaling, highlighting a novel biomarker-informed treatment strategy for prostate cancer.
©2023 American Association for Cancer Research.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
