

AML with complex karyotype: extreme genomic complexity revealed by combined long-read sequencing and Hi-C technology
- PMID: 37582288
- PMCID: PMC10632680
- DOI: 10.1182/bloodadvances.2023010887
AML with complex karyotype: extreme genomic complexity revealed by combined long-read sequencing and Hi-C technology
Abstract
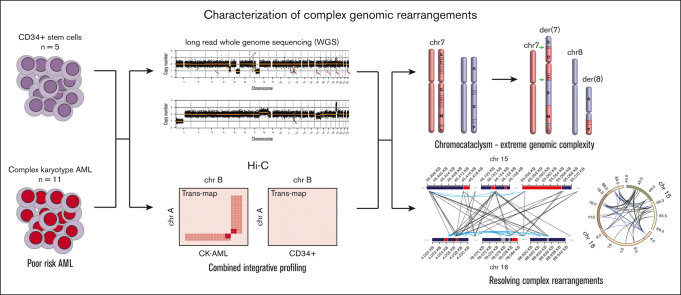
Acute myeloid leukemia with complex karyotype (CK-AML) is associated with poor prognosis, which is only in part explained by underlying TP53 mutations. Especially in the presence of complex chromosomal rearrangements, such as chromothripsis, the outcome of CK-AML is dismal. However, this degree of complexity of genomic rearrangements contributes to the leukemogenic phenotype and treatment resistance of CK-AML remains largely unknown. Applying an integrative workflow for the detection of structural variants (SVs) based on Oxford Nanopore (ONT) genomic DNA long-read sequencing (gDNA-LRS) and high-throughput chromosome confirmation capture (Hi-C) in a well-defined cohort of CK-AML identified regions with an extreme density of SVs. These rearrangements consisted to a large degree of focal amplifications enriched in the proximity of mammalian-wide interspersed repeat elements, which often result in oncogenic fusion transcripts, such as USP7::MVD, or the deregulation of oncogenic driver genes as confirmed by RNA-seq and ONT direct complementary DNA sequencing. We termed this novel phenomenon chromocataclysm. Thus, our integrative SV detection workflow combing gDNA-LRS and Hi-C enables to unravel complex genomic rearrangements at a very high resolution in regions hard to analyze by conventional sequencing technology, thereby providing an important tool to identify novel important drivers underlying cancer with complex karyotypic changes.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: L.B. has advisory role in AbbVie, Amgen, Astellas, Bristol Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Sanofi, and Seattle Genetics; and receives research funding from Bayer and Jazz Pharmaceuticals. The remaining authors declare no competing financial interests.
Figures





References
-
- Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70–87. - PubMed
-
- Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–1377. - PubMed
-
- Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
