

Single-cell multi-omics defines the cell-type-specific impact of splicing aberrations in human hematopoietic clonal outgrowths
- PMID: 37582363
- PMCID: PMC10528176
- DOI: 10.1016/j.stem.2023.07.012
Single-cell multi-omics defines the cell-type-specific impact of splicing aberrations in human hematopoietic clonal outgrowths
Abstract
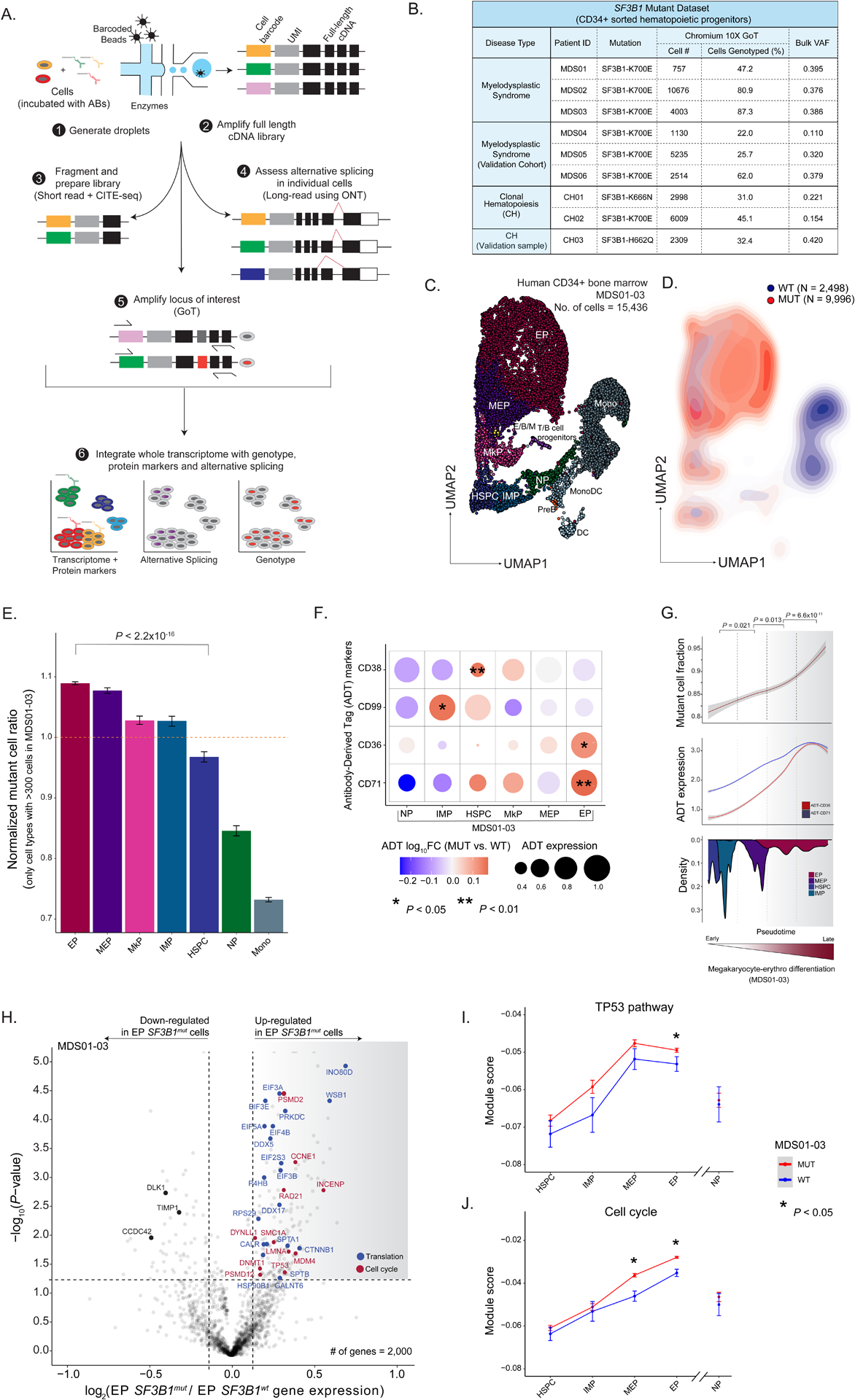
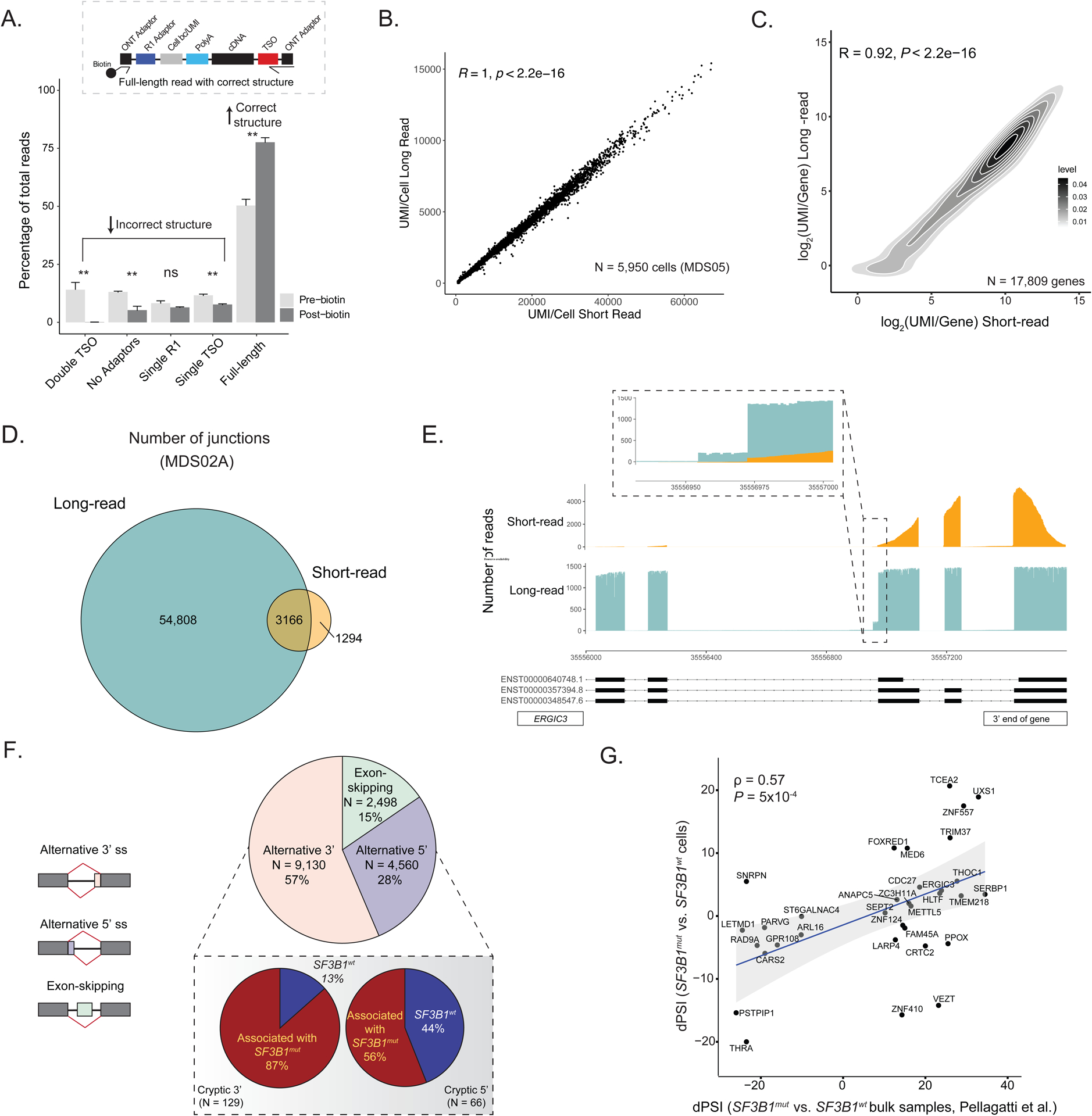
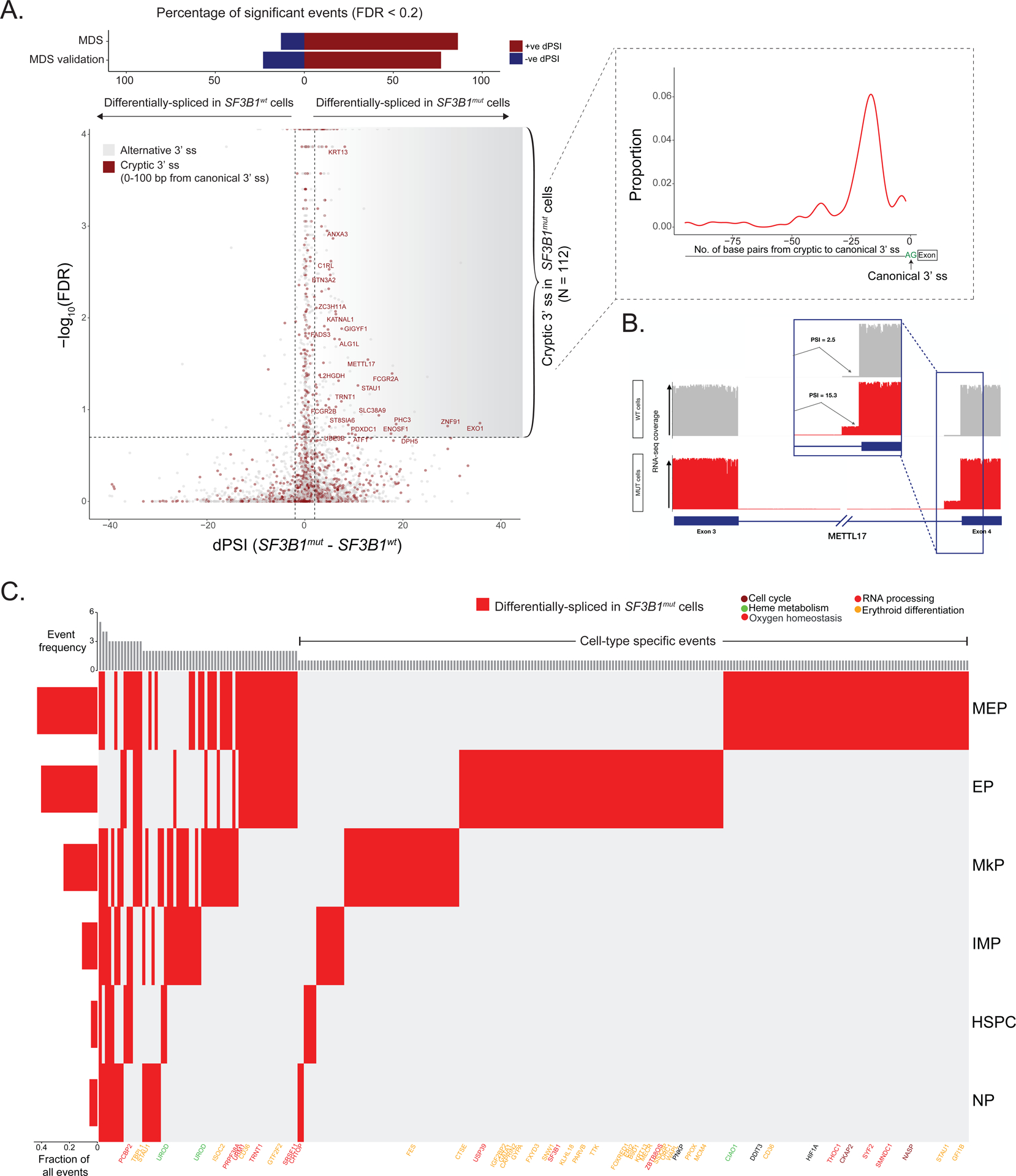
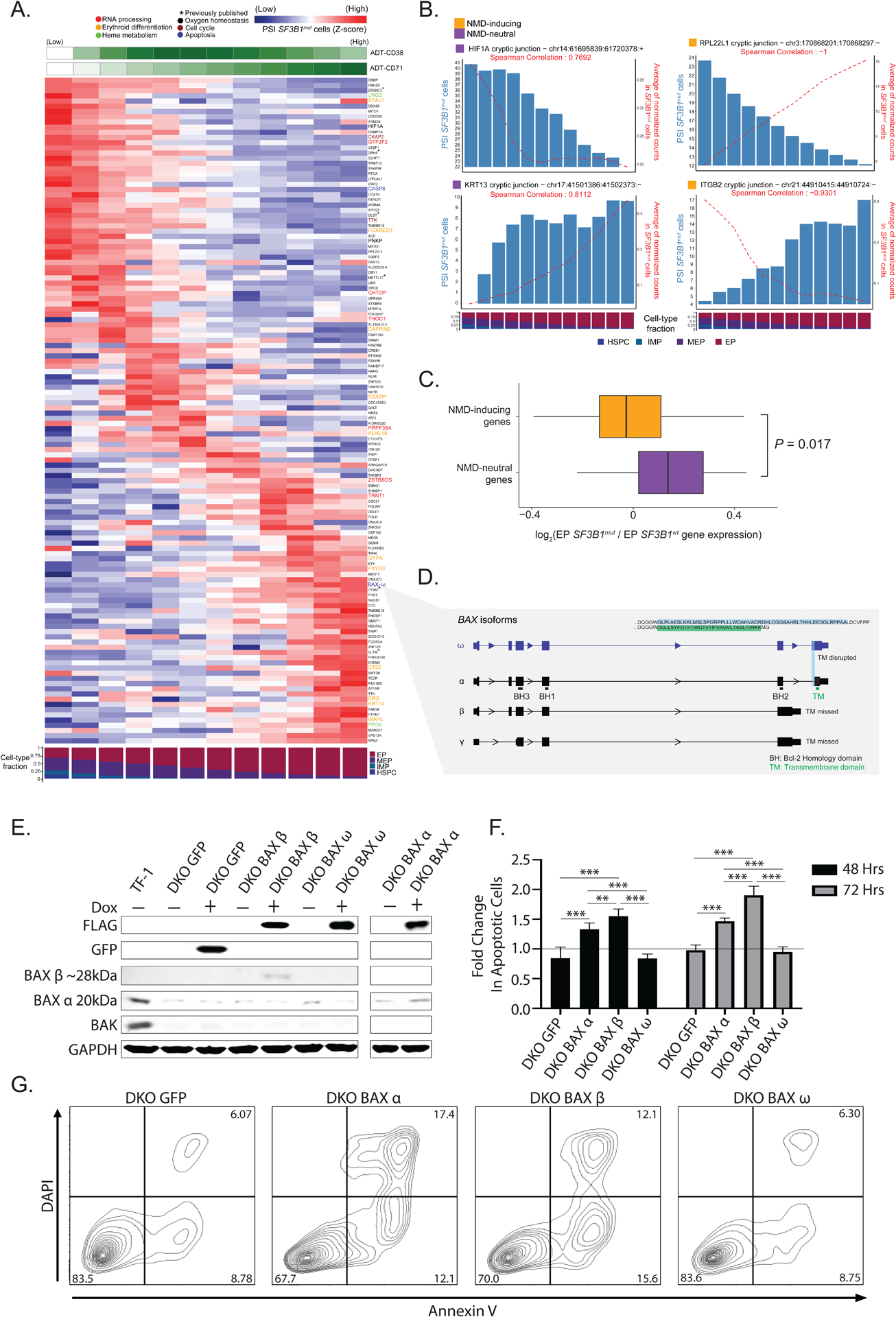
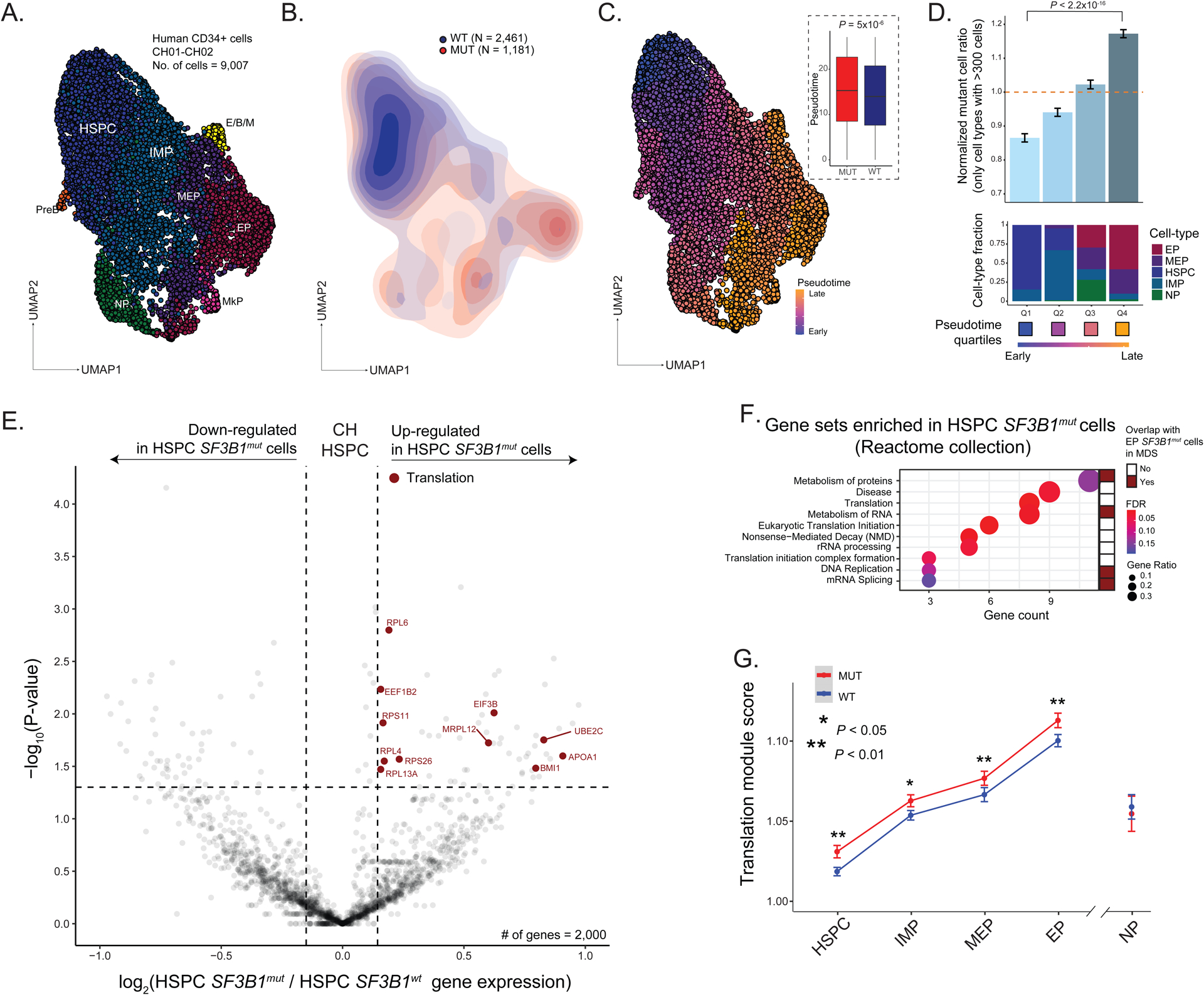
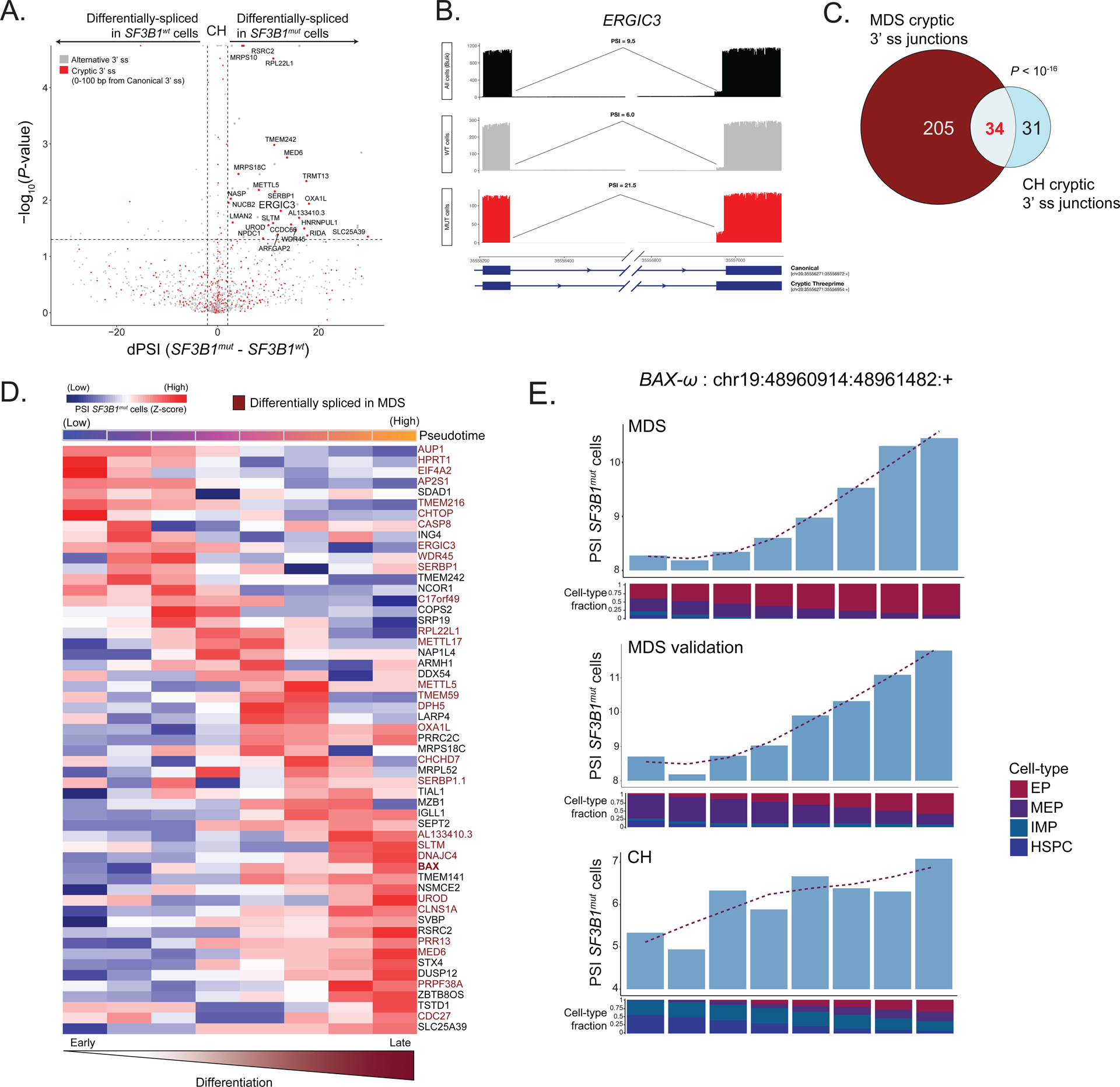
RNA splicing factors are recurrently mutated in clonal blood disorders, but the impact of dysregulated splicing in hematopoiesis remains unclear. To overcome technical limitations, we integrated genotyping of transcriptomes (GoT) with long-read single-cell transcriptomics and proteogenomics for single-cell profiling of transcriptomes, surface proteins, somatic mutations, and RNA splicing (GoT-Splice). We applied GoT-Splice to hematopoietic progenitors from myelodysplastic syndrome (MDS) patients with mutations in the core splicing factor SF3B1. SF3B1mut cells were enriched in the megakaryocytic-erythroid lineage, with expansion of SF3B1mut erythroid progenitor cells. We uncovered distinct cryptic 3' splice site usage in different progenitor populations and stage-specific aberrant splicing during erythroid differentiation. Profiling SF3B1-mutated clonal hematopoiesis samples revealed that erythroid bias and cell-type-specific cryptic 3' splice site usage in SF3B1mut cells precede overt MDS. Collectively, GoT-Splice defines the cell-type-specific impact of somatic mutations on RNA splicing, from early clonal outgrowths to overt neoplasia, directly in human samples.
Keywords: BAX; RNA-seq; SF3B1; clonal hematopoiesis; genotyping; long-read sequencing; multi-omics; myelodysplastic syndrome; single cell; splicing.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.G. serves as a consultant for S2 Genomics Inc. X.D., J.B., A.W.D., S.H., S.J., and E.H. are employees of Oxford Nanopore Technologies Inc. and are shareholders and/or share option holders. I.M.G. serves on the advisory or consulting board of Bristol Myers Squibb, Takeda, Janssen, Sanofi, Novartis, Amgen, Celgene, Cellectar, Pfizer, Menarini Silicon Biosystems, Oncopeptides, The Binding Site, GlazoSmithKlein, AbbVie, Adaptive, and 10x Genomics. O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc., Merck, Pfizer, and Janssen, and O.A.-W. is on the Scientific Advisory Board of Envisagenics Inc. and AIChemy. O.A.-W. has received prior research funding from H3B Biomedicine and LOXO Oncology unrelated to the current manuscript. D.A.L. has served as a consultant for AbbVie, AstraZeneca, and Illumina and is on the Scientific Advisory Board of Mission Bio, Pangea, Alethiomics, and C2i Genomics; D.A.L. has received prior research funding from BMS, 10x Genomics, Ultima Genomics, and Illumina unrelated to the current manuscript.
Figures
References
-
- Teixeira VH, Pipinikas CP, Pennycuick A, Lee-Six H, Chandrasekharan D, Beane J, Morris TJ, Karpathakis A, Feber A, Breeze CE, et al. (2019). Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat Med 25, 517–525. 10.1038/s41591-018-0323-0. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
