

Genome sequencing of 2000 canids by the Dog10K consortium advances the understanding of demography, genome function and architecture
- PMID: 37582787
- PMCID: PMC10426128
- DOI: 10.1186/s13059-023-03023-7
Genome sequencing of 2000 canids by the Dog10K consortium advances the understanding of demography, genome function and architecture
Erratum in
-
Author Correction: Genome sequencing of 2000 canids by the Dog10K consortium advances the understanding of demography, genome function and architecture.Genome Biol. 2023 Nov 7;24(1):255. doi: 10.1186/s13059-023-03101-w. Genome Biol. 2023. PMID: 37936157 Free PMC article. No abstract available.
Abstract
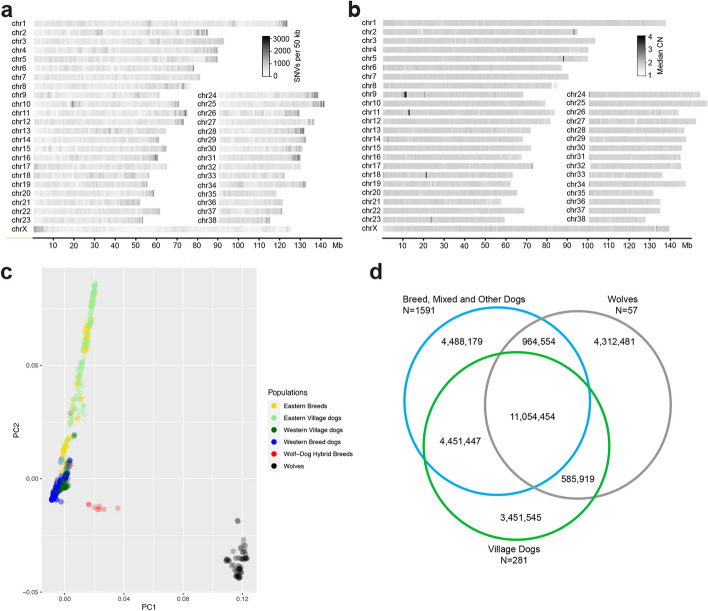
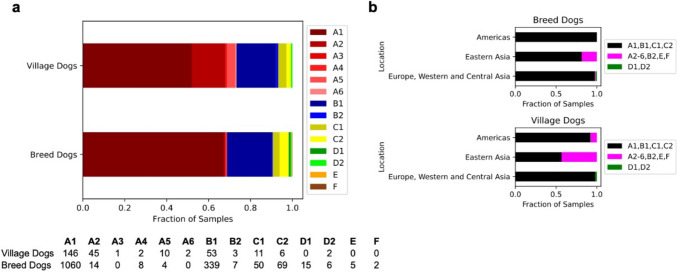
Background: The international Dog10K project aims to sequence and analyze several thousand canine genomes. Incorporating 20 × data from 1987 individuals, including 1611 dogs (321 breeds), 309 village dogs, 63 wolves, and four coyotes, we identify genomic variation across the canid family, setting the stage for detailed studies of domestication, behavior, morphology, disease susceptibility, and genome architecture and function.
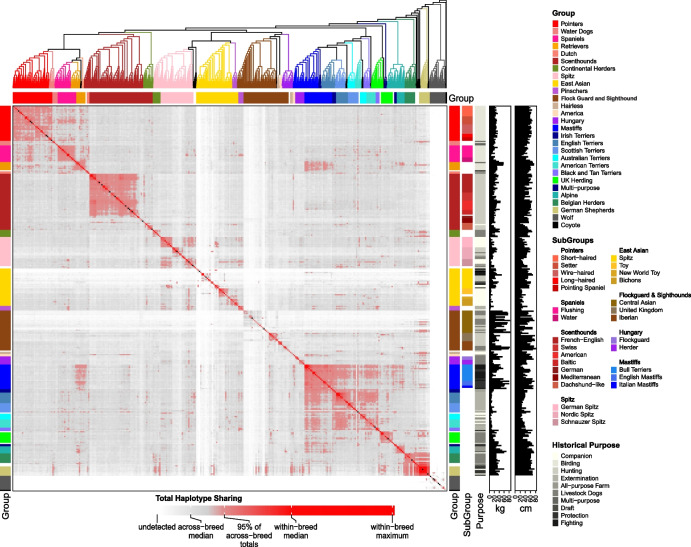
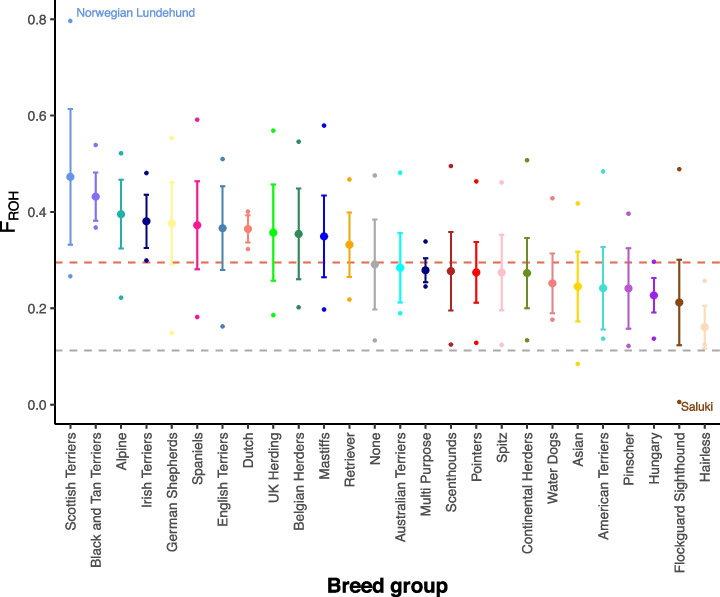
Results: We report the analysis of > 48 M single-nucleotide, indel, and structural variants spanning the autosomes, X chromosome, and mitochondria. We discover more than 75% of variation for 239 sampled breeds. Allele sharing analysis indicates that 94.9% of breeds form monophyletic clusters and 25 major clades. German Shepherd Dogs and related breeds show the highest allele sharing with independent breeds from multiple clades. On average, each breed dog differs from the UU_Cfam_GSD_1.0 reference at 26,960 deletions and 14,034 insertions greater than 50 bp, with wolves having 14% more variants. Discovered variants include retrogene insertions from 926 parent genes. To aid functional prioritization, single-nucleotide variants were annotated with SnpEff and Zoonomia phyloP constraint scores. Constrained positions were negatively correlated with allele frequency. Finally, the utility of the Dog10K data as an imputation reference panel is assessed, generating high-confidence calls across varied genotyping platform densities including for breeds not included in the Dog10K collection.
Conclusions: We have developed a dense dataset of 1987 sequenced canids that reveals patterns of allele sharing, identifies likely functional variants, informs breed structure, and enables accurate imputation. Dog10K data are publicly available.
Keywords: Canine; Demographic history; Dog; Genetic diversity; Genomics; Mitochondrial DNA; Variation.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
BWD is Director of Veterinary Research at Volition Veterinary LLC; ARB is a Co-Founder and CSO of Embark Veterinary Inc. and serves on the Board of EMBARK and the Morris Animal Foundation. HL has consulted with Wisdom Panel Kinship, is an owner and chairman of the board of Petbiomics Ltd., and Petmeta Labs Ltd., and is an owner and advisor of DeepScan Dignostics Ltd. IT is the curator of Online Mendelian Inheritance which is supported by the University of Sydney.
Figures







References
-
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, 3rd, Zody MC, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. - PubMed
