

The ever wider clinical spectrum of RMND1-related disorders and limitedness of phenotype-based classifications
- PMID: 37584739
- PMCID: PMC10560146
- DOI: 10.1007/s00109-023-02356-x
The ever wider clinical spectrum of RMND1-related disorders and limitedness of phenotype-based classifications
Abstract
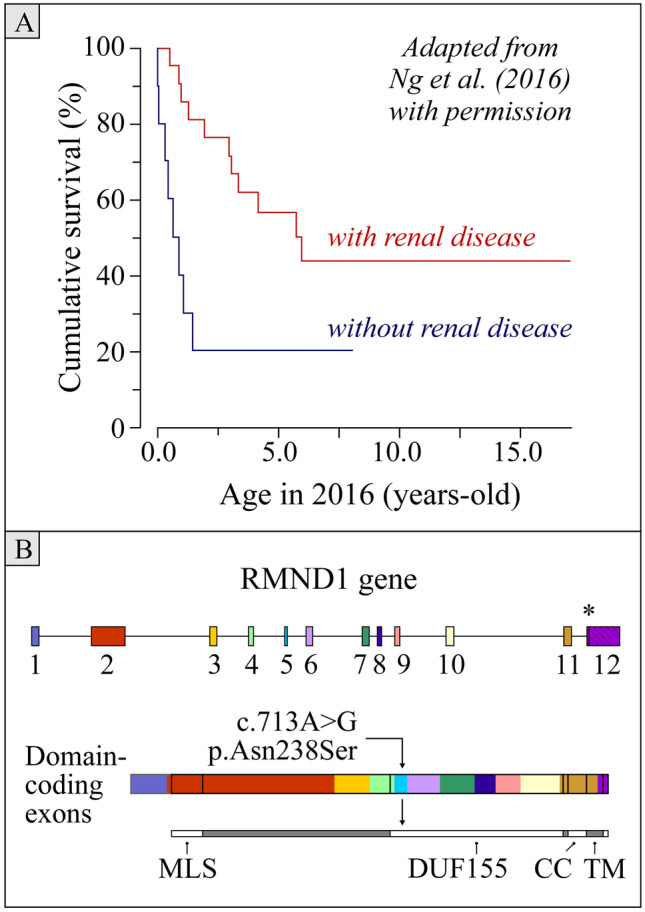
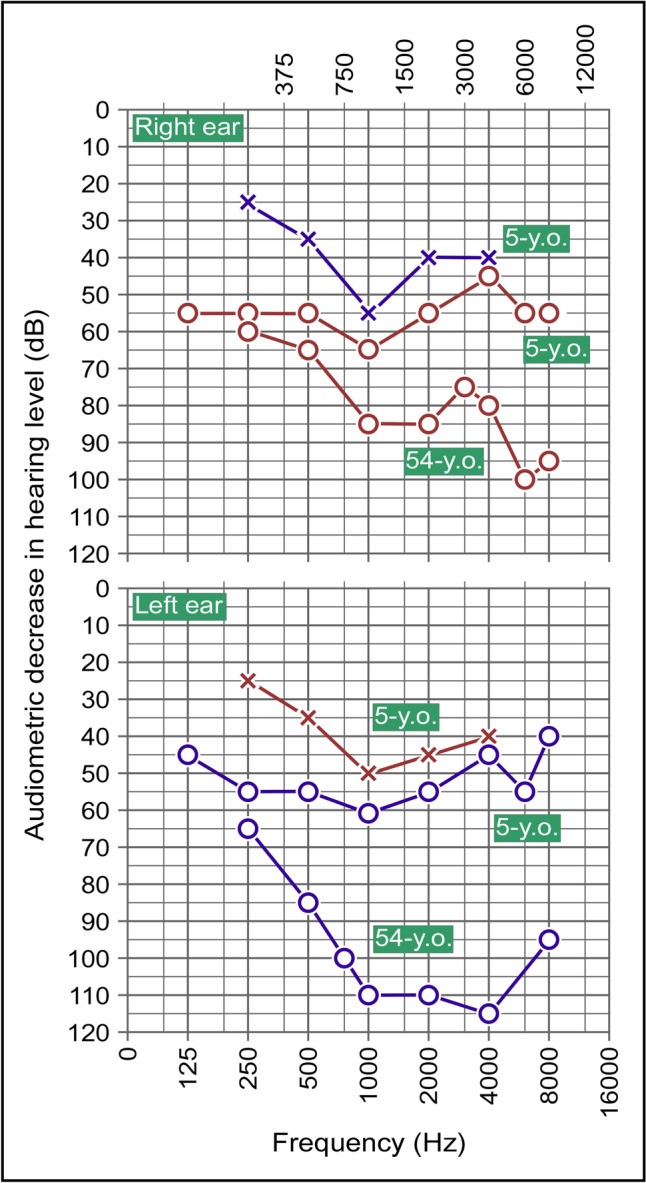
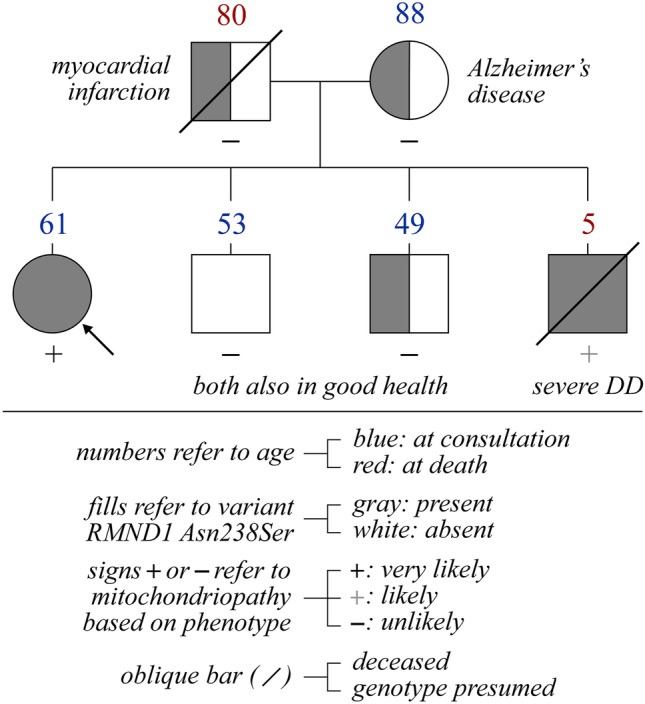
RMND1 has been identified as a mitochondriopathy-associated gene less than 12 years ago. The most common phenotype related to this gene is an early onset, severe form of encephalomyopathy that leads to death in a medium time of three years after birth. However, milder and later onset presentations have been reported in some individuals, including two in whom the mitochondriopathy was identified at ~ 40 years of age, and the early onset presentations have been the object of no reports in those who survived beyond age 10. It is thus unclear how lethal RMND1-related conditions really are. We herein describe the oldest case to have been identified hitherto with this condition, i.e., that of a white female who was 61 at the time of diagnosis but was still active in her everyday life. The gene defect identified was nonetheless associated with many manifestations including ovarian insufficiency and sensorineural hearing loss (two features of what is currently designated as Perrault syndrome) as well as chronic renal failure, asymptomatic myopathy, leukopenia, and a few others. In our opinion, this case is of great translational interest for at least three reasons. First, it hints towards the possibility of near-normal life expectancies in some if not many individuals with RMND1 insufficiency. Second, it underlines the wide clinical spectrum associated with this gene. Third, it brings us to question the use of eponyms and syndromic features to identify the true etiology of multisystemic phenotypes. KEY MESSAGES: RMND1-related conditions typically manifest at an early age with a progressive and lethal form of encephalomyopathy. More benign presentations have been described with some being categorized as Perrault syndrome but none have been diagnosed after the age of 45. The clinical spectrum and presenting age of RMND1-related mitochondriopathies are probably much more varied than implied in the current literature. The case reported in this manuscript illustrates the limitedness of phenotype-based classifications of genetic disorders to identify the defect at cause.
Keywords: Mitochondriopathy; Myelodysplasia; Myopathy; Perrault syndrome; RMND1.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Janer A, Antonicka H, Lalonde E, Nishimura T, Sasarman F, Brown GK, Brown RM, Majewski J, Shoubridge EA. An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet. 2012;91:737–743. doi: 10.1016/j.ajhg.2012.08.020. - DOI - PMC - PubMed
-
- Garcia-Diaz B, Barros MH, Sanna-Cherchi S, Emmanuele V, Akman HO, Ferreiro-Barros CC, Horvath R, Tadesse S, El Gharaby N, DiMauro S, et al. Infantile encephaloneuromyopathy and defective mitochondrial translation are due to a homozygous RMND1 mutation. Am J Hum Genet. 2012;91:729–736. doi: 10.1016/j.ajhg.2012.08.019. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
