

Management of adults with Alagille syndrome
- PMID: 37584849
- PMCID: PMC10522532
- DOI: 10.1007/s12072-023-10578-x
Management of adults with Alagille syndrome
Abstract
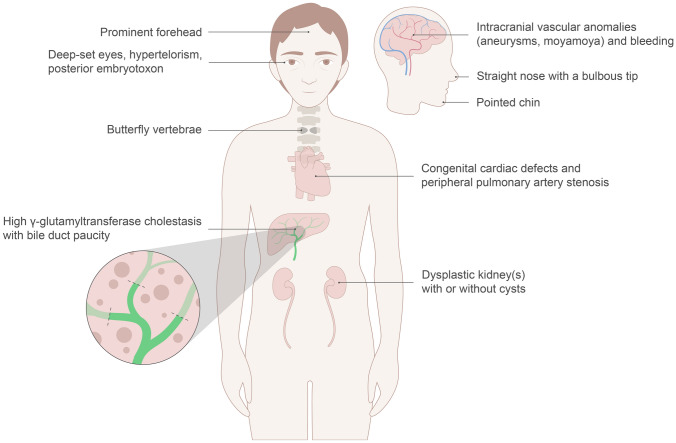
Alagille syndrome (ALGS) is a complex rare genetic disorder that involves multiple organ systems and is historically regarded as a disease of childhood. Since it is inherited in an autosomal dominant manner in 40% of patients, it carries many implications for genetic counselling of patients and screening of family members. In addition, the considerable variable expression and absence of a clear genotype-phenotype correlation, results in a diverse range of clinical manifestations, even in affected individuals within the same family. With recent therapeutic advancements in cholestasis treatment and the improved survival rates with liver transplantation (LT), many patients with ALGS survive into adulthood. Although LT is curative for liver disease secondary to ALGS, complications secondary to extrahepatic involvement remain problematic lifelong. This review is aimed at providing a comprehensive review of ALGS to adult clinicians who will take over the medical care of these patients following transition, with particular focus on certain aspects of the condition that require lifelong surveillance. We also provide a diagnostic framework for adult patients with suspected ALGS and highlight key aspects to consider when determining eligibility for LT in patients with this syndrome.
Keywords: Bile duct paucity; Cholestatic liver disease; Hepatocellular carcinoma; JAG1; Liver transplant; Maralixibat; NOTCH2; Portal hypertension; Pruritus; Regenerative nodules.
© 2023. The Author(s).
Conflict of interest statement
MDA, AAB, and VK declare they have no financial interests to declare. SMV is a consultant for Mirum and Albireo. BMK is a consultant for Mirum, Albireo, and Audentes (Astellas). BMK received unrestricted educational grant funding from Mirum and Albireo.
Figures
References
-
- Alagille D, Habib E, Thomassin N. L'atresie des voies biliaires intrahepatiques avec voies biliaires extrahepatiques permeables chez l'enfant. J Par Pediatr. 1969;301:301–318. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
