

Personalized neoantigen viro-immunotherapy platform for triple-negative breast cancer
- PMID: 37586771
- PMCID: PMC10432671
- DOI: 10.1136/jitc-2023-007336
Personalized neoantigen viro-immunotherapy platform for triple-negative breast cancer
Abstract
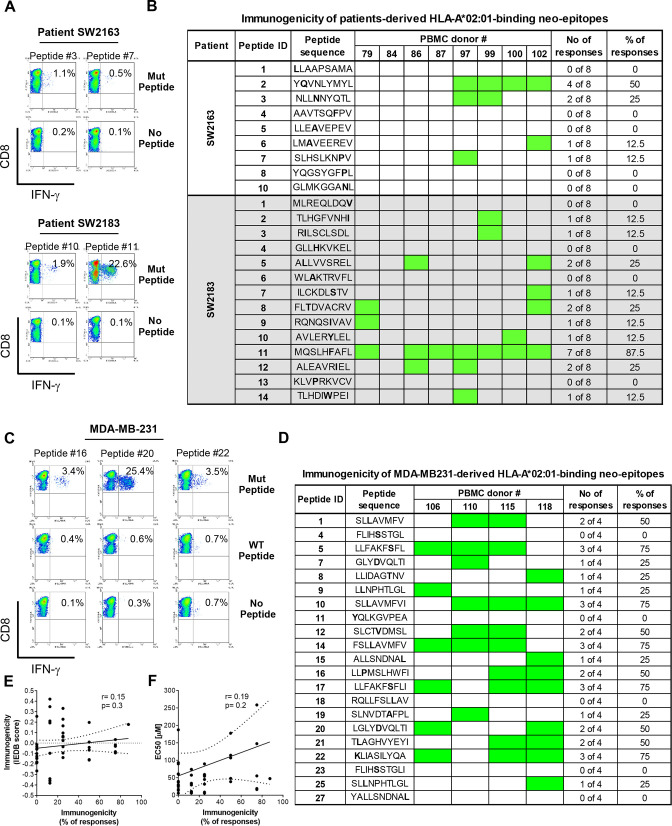
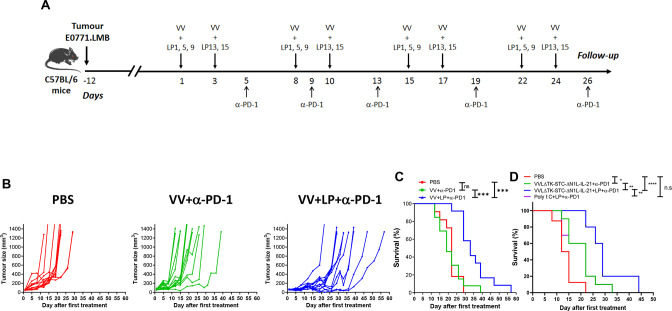
Background: Triple-negative breast cancer (TNBC) corresponds to approximately 20% of all breast tumors, with a high propensity for metastasis and a poor prognosis. Because TNBC displays a high mutational load compared with other breast cancer types, a neoantigen-based immunotherapy strategy could be effective. One major bottleneck in the development of a neoantigen-based vaccine for TNBC is the selection of the best targets, that is, tumor-specific neoantigens which are presented at the surface of tumor cells and capable of eliciting robust immune responses. In this study, we aimed to set up a platform for identification and delivery of immunogenic neoantigens in a vaccine regimen for TNBC using oncolytic vaccinia virus (VV).
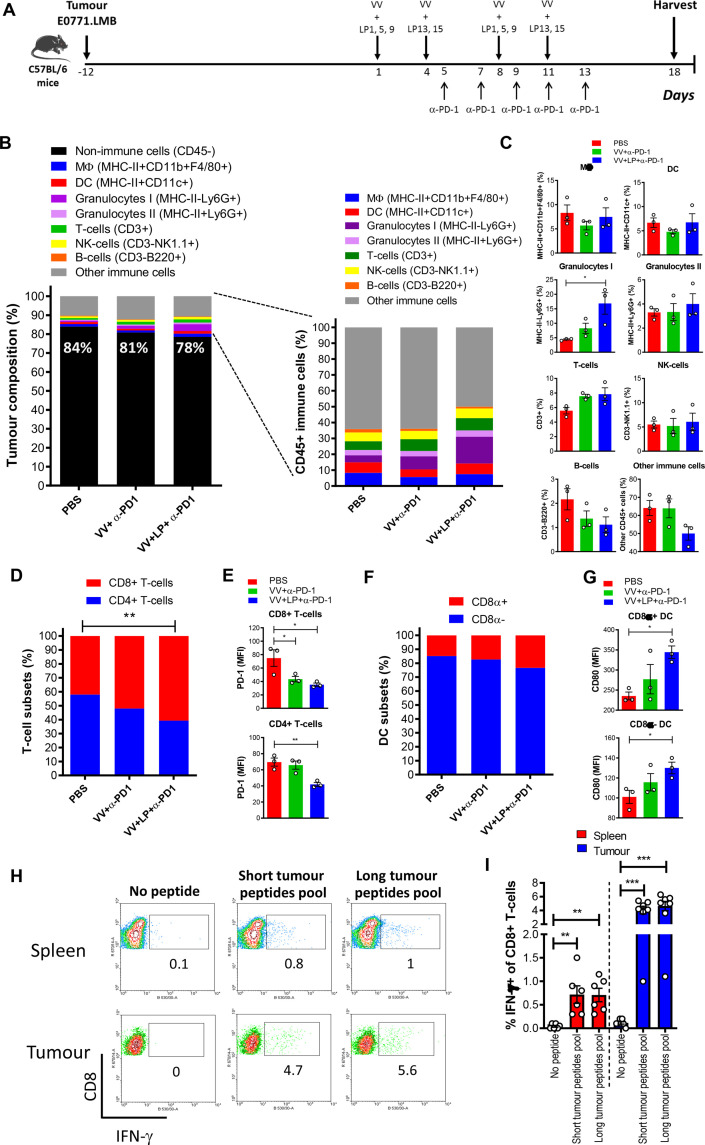
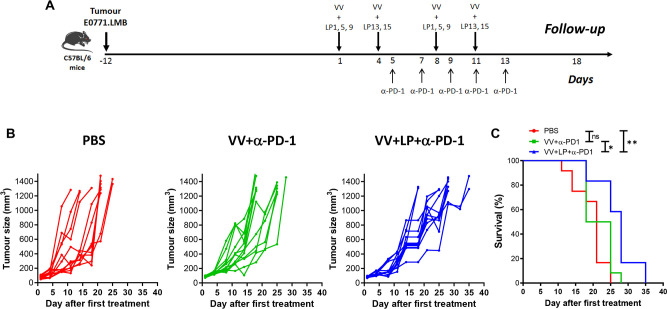
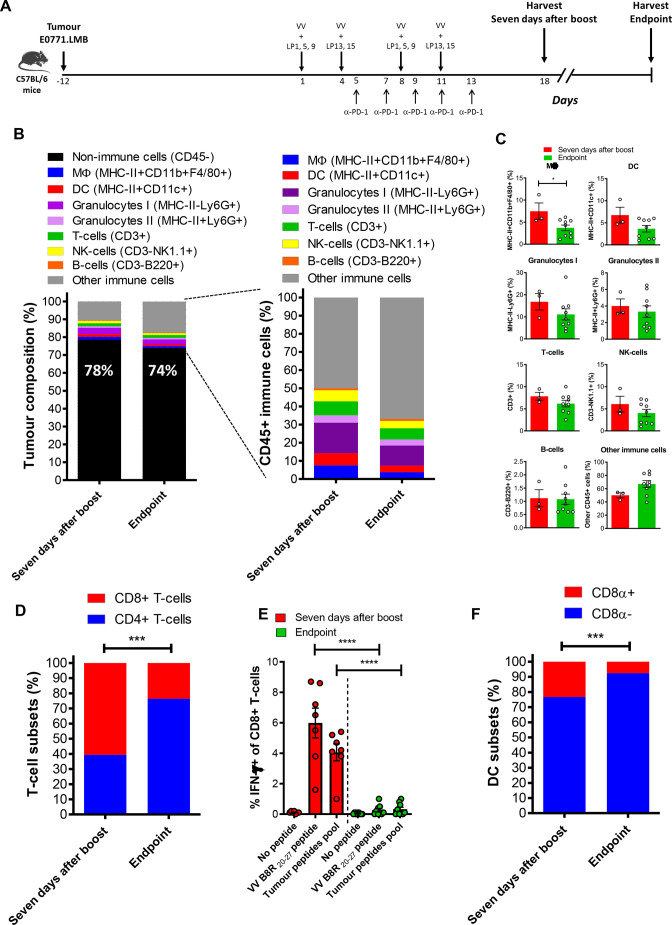
Methods: We used bioinformatic tools and cell-based assays to identify immunogenic neoantigens in TNBC patients' samples, human and murine cell lines. Immunogenicity of the neoantigens was tested in vitro (human) and ex vivo (murine) in T-cell assays. To assess the efficacy of our regimen, we used a preclinical model of TNBC where we treated tumor-bearing mice with neoantigens together with oncolytic VV and evaluated the effect on induction of neoantigen-specific CD8+T cells, tumor growth and survival.
Results: We successfully identified immunogenic neoantigens and generated neoantigen-specific CD8+T cells capable of recognizing a human TNBC cell line expressing the mutated gene. Using a preclinical model of TNBC, we showed that our tumor-specific oncolytic VV was able to change the tumor microenvironment, attracting and maintaining mature cross-presenting CD8α+dendritic cells and effector T-cells. Moreover, when delivered in a prime/boost regimen together with oncolytic VV, long peptides encompassing neoantigens were able to induce neoantigen-specific CD8+T cells, slow tumor growth and increase survival.
Conclusions: Our study provides a promising approach for the development of neoantigen-based immunotherapies for TNBC. By identifying immunogenic neoantigens and developing a delivery system through tumor-specific oncolytic VV, we have demonstrated that neoantigen-based vaccines could be effective in inducing neoantigen-specific CD8+T cells response with significant impact on tumor growth. Further studies are needed to determine the safety and efficacy of this approach in clinical trials.
Keywords: Breast Neoplasms; Immunogenicity, Vaccine; Immunotherapy; Oncolytic Virotherapy.
© Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials