

Transient naive reprogramming corrects hiPS cells functionally and epigenetically
- PMID: 37587336
- PMCID: PMC10447250
- DOI: 10.1038/s41586-023-06424-7
Transient naive reprogramming corrects hiPS cells functionally and epigenetically
Abstract
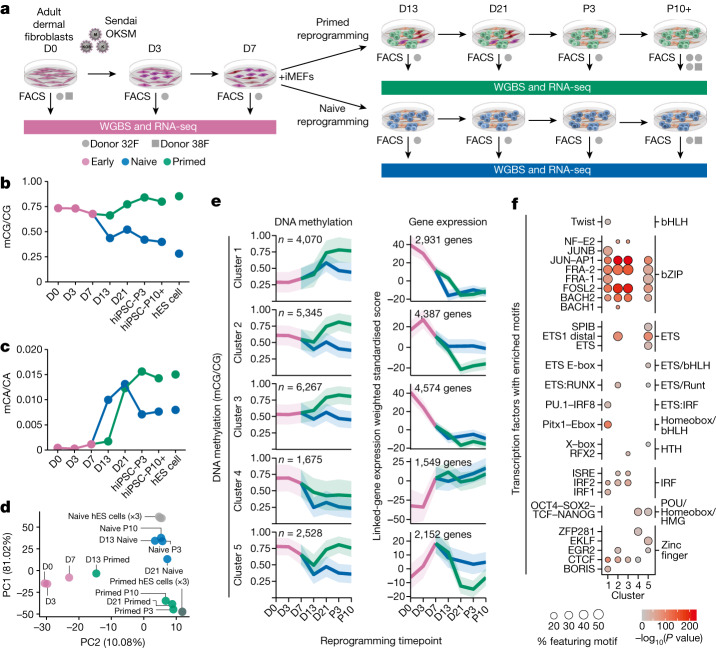
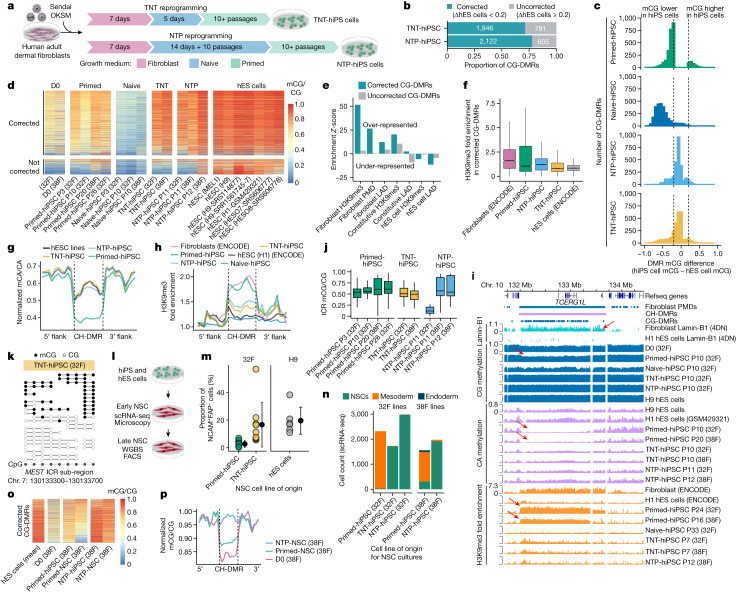
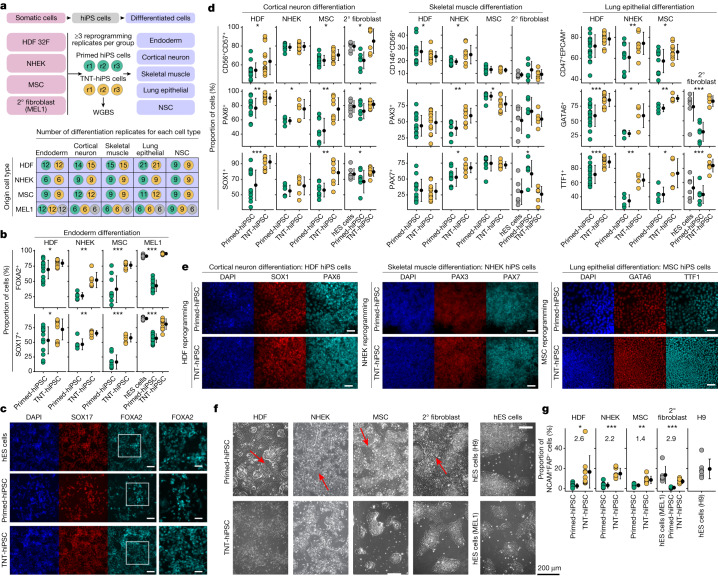
Cells undergo a major epigenome reconfiguration when reprogrammed to human induced pluripotent stem cells (hiPS cells). However, the epigenomes of hiPS cells and human embryonic stem (hES) cells differ significantly, which affects hiPS cell function1-8. These differences include epigenetic memory and aberrations that emerge during reprogramming, for which the mechanisms remain unknown. Here we characterized the persistence and emergence of these epigenetic differences by performing genome-wide DNA methylation profiling throughout primed and naive reprogramming of human somatic cells to hiPS cells. We found that reprogramming-induced epigenetic aberrations emerge midway through primed reprogramming, whereas DNA demethylation begins early in naive reprogramming. Using this knowledge, we developed a transient-naive-treatment (TNT) reprogramming strategy that emulates the embryonic epigenetic reset. We show that the epigenetic memory in hiPS cells is concentrated in cell of origin-dependent repressive chromatin marked by H3K9me3, lamin-B1 and aberrant CpH methylation. TNT reprogramming reconfigures these domains to a hES cell-like state and does not disrupt genomic imprinting. Using an isogenic system, we demonstrate that TNT reprogramming can correct the transposable element overexpression and differential gene expression seen in conventional hiPS cells, and that TNT-reprogrammed hiPS and hES cells show similar differentiation efficiencies. Moreover, TNT reprogramming enhances the differentiation of hiPS cells derived from multiple cell types. Thus, TNT reprogramming corrects epigenetic memory and aberrations, producing hiPS cells that are molecularly and functionally more similar to hES cells than conventional hiPS cells. We foresee TNT reprogramming becoming a new standard for biomedical and therapeutic applications and providing a novel system for studying epigenetic memory.
© 2023. The Author(s).
Conflict of interest statement
S.B., X.L., J. M. Polo and R.L. are co-inventors on a pending patent (PCT/AU2019/051296) filed by the University of Western Australia and Monash University related to this work. R.L. is a co-inventor on a patent (WO/2012/058634) concerning methods of characterizing the epigenetic signature of human induced pluripotent stem cells. Although unrelated to this manuscript, O.J.L.R. and J. M. Polo are co-inventors on a patent (WO/2017/106932) and are co-founders and shareholders of Mogrify, a cell therapy company. X.L. is a co-founder of iCamuno Biotherapeutics. The other authors declare no competing interests.
Figures












References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
