

Nuclear genetic control of mtDNA copy number and heteroplasmy in humans
- PMID: 37587338
- PMCID: PMC10447254
- DOI: 10.1038/s41586-023-06426-5
Nuclear genetic control of mtDNA copy number and heteroplasmy in humans
Erratum in
-
Author Correction: Nuclear genetic control of mtDNA copy number and heteroplasmy in humans.Nature. 2024 Jun;630(8017):E10. doi: 10.1038/s41586-024-07364-6. Nature. 2024. PMID: 38831054 Free PMC article. No abstract available.
Abstract
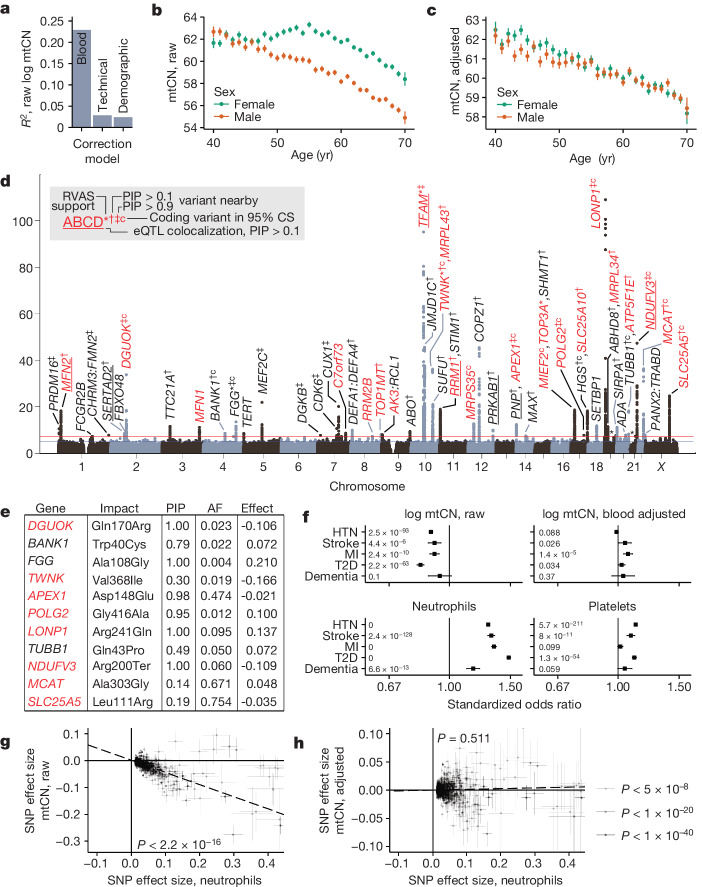
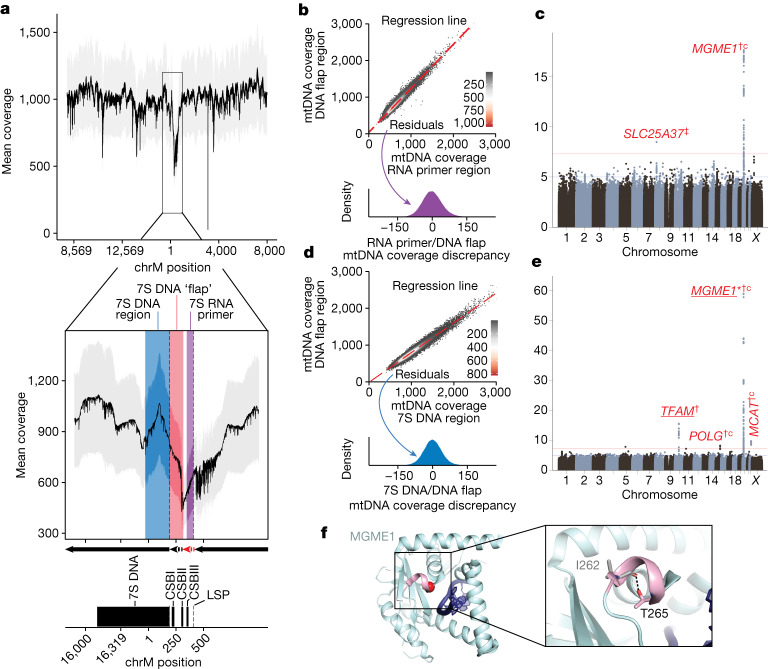
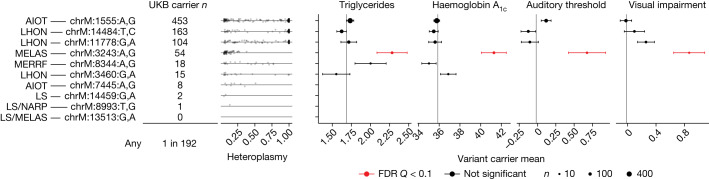
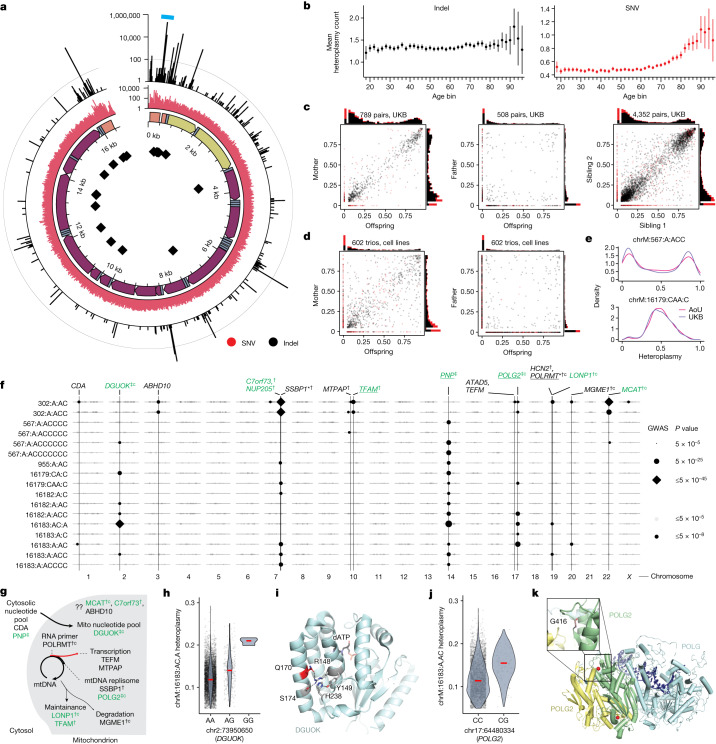
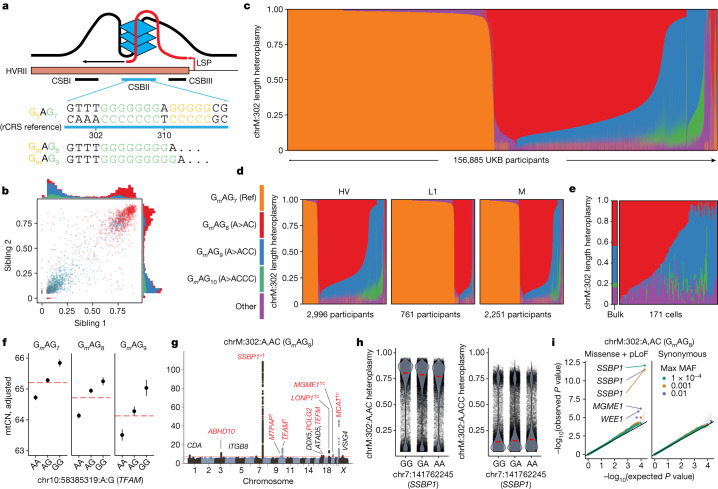
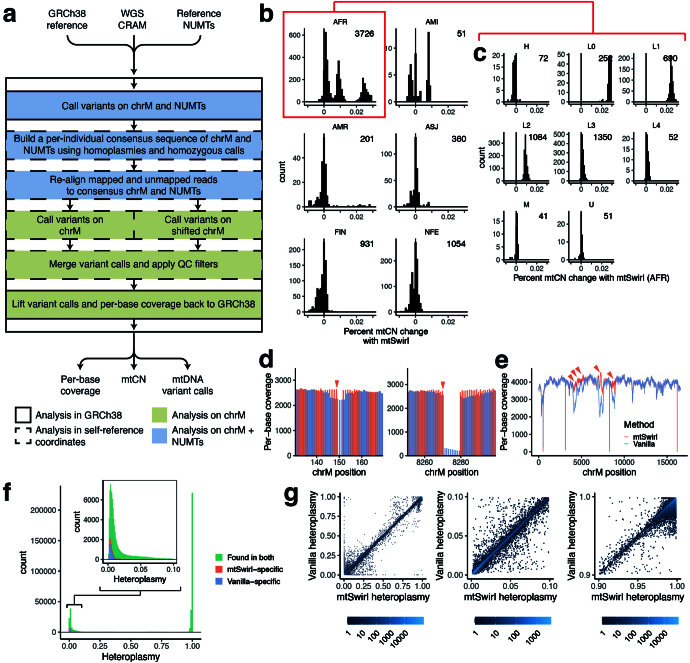
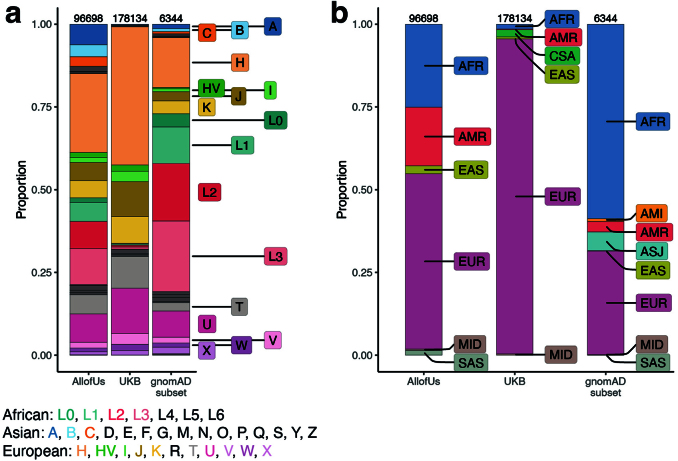
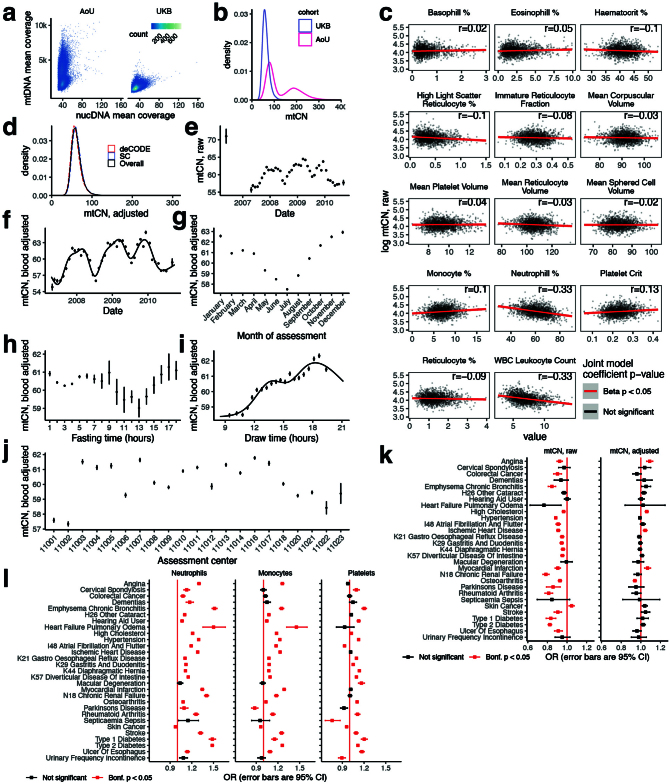
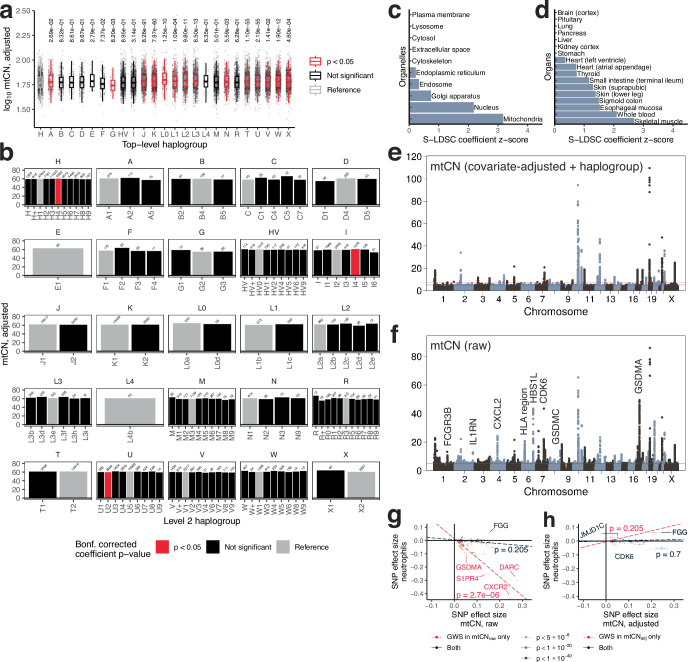
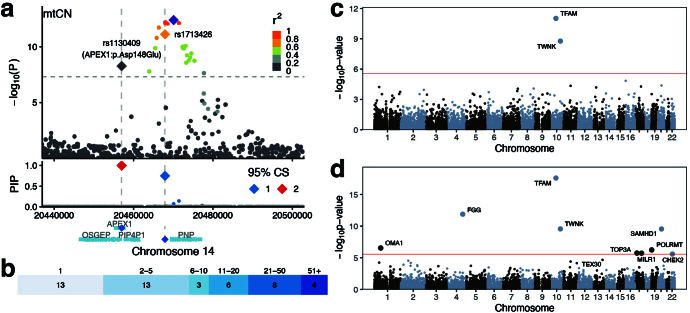
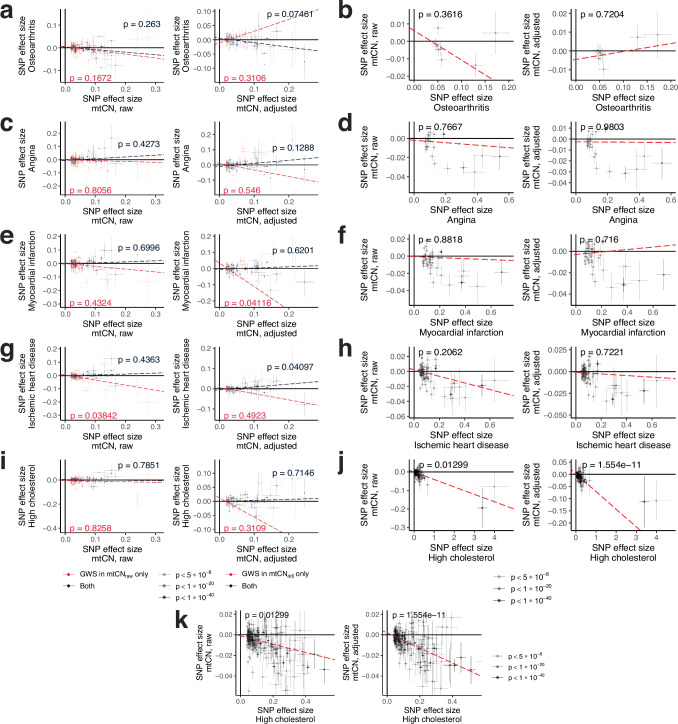
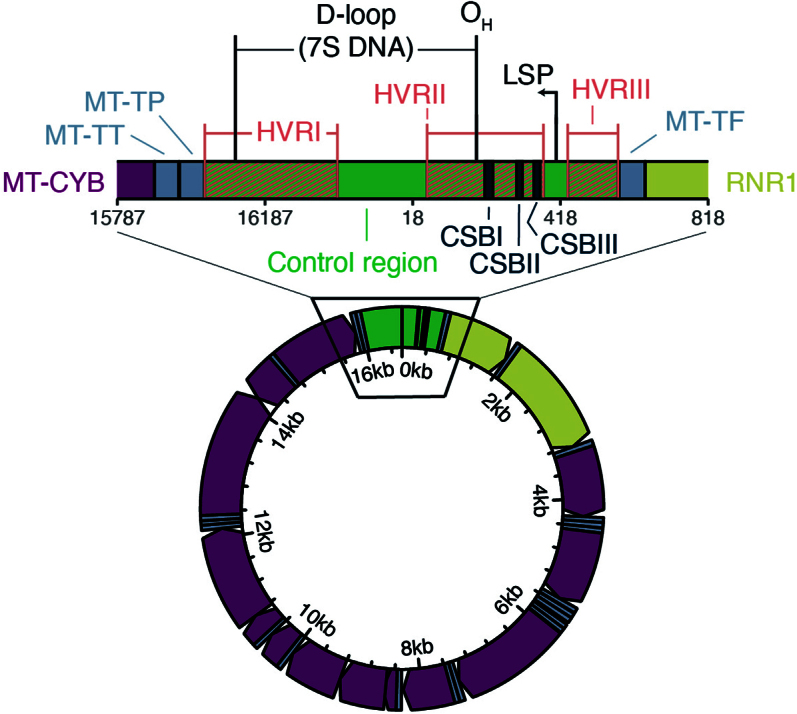
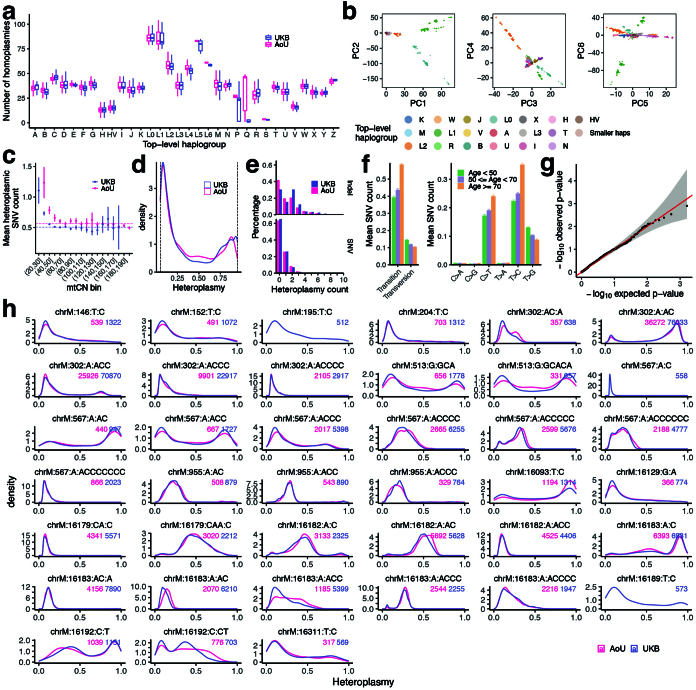
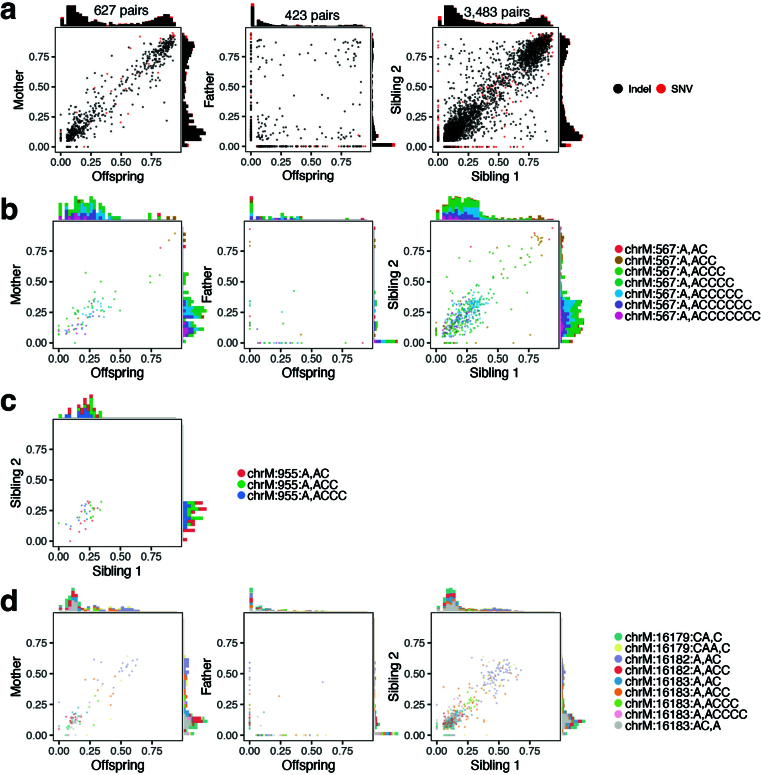
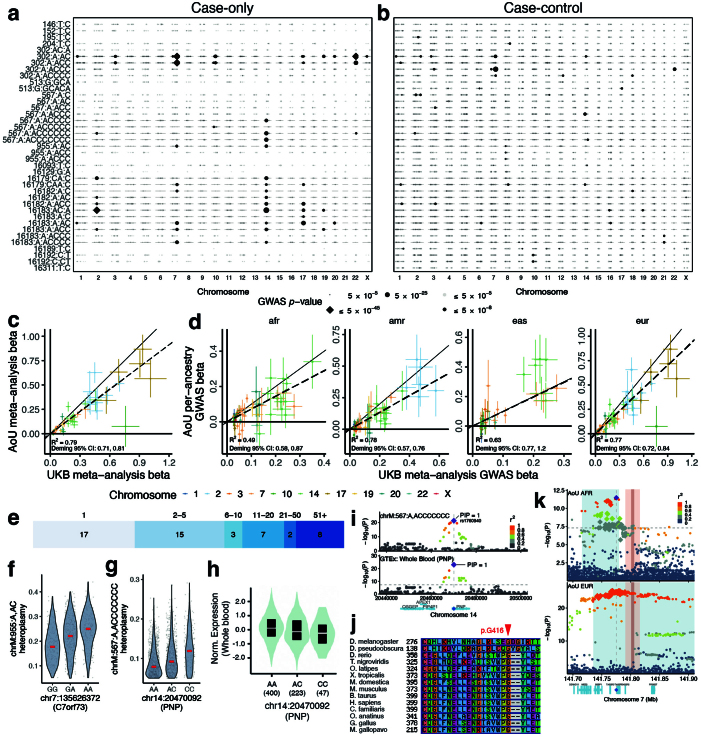
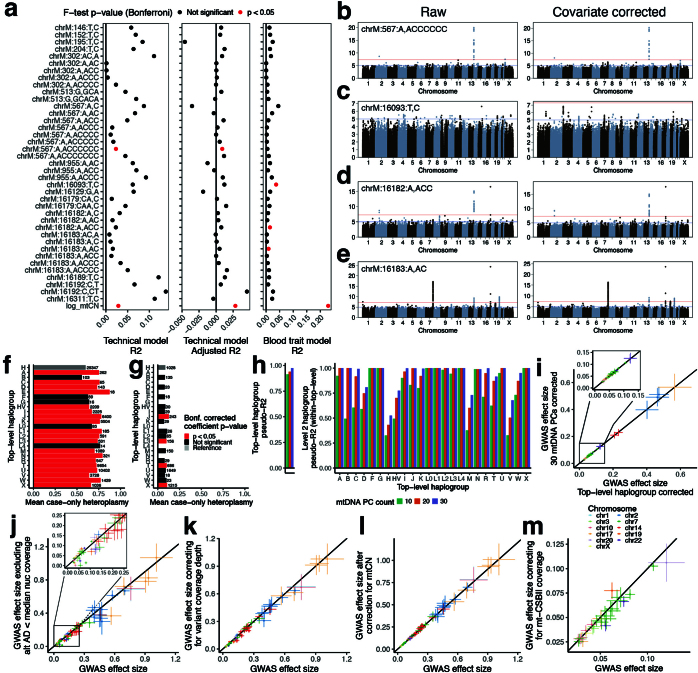
Mitochondrial DNA (mtDNA) is a maternally inherited, high-copy-number genome required for oxidative phosphorylation1. Heteroplasmy refers to the presence of a mixture of mtDNA alleles in an individual and has been associated with disease and ageing. Mechanisms underlying common variation in human heteroplasmy, and the influence of the nuclear genome on this variation, remain insufficiently explored. Here we quantify mtDNA copy number (mtCN) and heteroplasmy using blood-derived whole-genome sequences from 274,832 individuals and perform genome-wide association studies to identify associated nuclear loci. Following blood cell composition correction, we find that mtCN declines linearly with age and is associated with variants at 92 nuclear loci. We observe that nearly everyone harbours heteroplasmic mtDNA variants obeying two principles: (1) heteroplasmic single nucleotide variants tend to arise somatically and accumulate sharply after the age of 70 years, whereas (2) heteroplasmic indels are maternally inherited as mixtures with relative levels associated with 42 nuclear loci involved in mtDNA replication, maintenance and novel pathways. These loci may act by conferring a replicative advantage to certain mtDNA alleles. As an illustrative example, we identify a length variant carried by more than 50% of humans at position chrM:302 within a G-quadruplex previously proposed to mediate mtDNA transcription/replication switching2,3. We find that this variant exerts cis-acting genetic control over mtDNA abundance and is itself associated in-trans with nuclear loci encoding machinery for this regulatory switch. Our study suggests that common variation in the nuclear genome can shape variation in mtCN and heteroplasmy dynamics across the human population.
© 2023. The Author(s).
Conflict of interest statement
V.K.M. is a paid advisor to 5am Ventures. B.M.N. is a member of the scientific advisory board at Deep Genomics and Neumora and a consultant of the scientific advisory board for Camp4 Therapeutics. K.J.K. is a consultant for Vor Biopharma. The remaining authors declare no competing interests.
Figures
Update of
-
Nuclear genetic control of mtDNA copy number and heteroplasmy in humans.medRxiv [Preprint]. 2023 Jan 19:2023.01.19.23284696. doi: 10.1101/2023.01.19.23284696. medRxiv. 2023. Update in: Nature. 2023 Aug;620(7975):839-848. doi: 10.1038/s41586-023-06426-5. PMID: 36711677 Free PMC article. Updated. Preprint.
Comment in
-
Mitochondrial DNA comes with nuclear strings attached.Nature. 2023 Aug;620(7975):732-734. doi: 10.1038/d41586-023-01783-7. Nature. 2023. PMID: 37587274 No abstract available.
References
MeSH terms
Substances
Grants and funding
- OT2 OD026556/OD/NIH HHS/United States
- U2C OD023196/OD/NIH HHS/United States
- F30 AG074507/AG/NIA NIH HHS/United States
- OT2 OD025315/OD/NIH HHS/United States
- OT2 OD026551/OD/NIH HHS/United States
- U24 OD023121/OD/NIH HHS/United States
- OT2 OD026549/OD/NIH HHS/United States
- MC_UU_00028/7/MRC_/Medical Research Council/United Kingdom
- OT2 OD026555/OD/NIH HHS/United States
- OT2 OD026553/OD/NIH HHS/United States
- OT2 OD025276/OD/NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- OT2 OD026554/OD/NIH HHS/United States
- U24 OD023163/OD/NIH HHS/United States
- U24 OD023176/OD/NIH HHS/United States
- OT2 OD026548/OD/NIH HHS/United States
- R37 MH107649/MH/NIMH NIH HHS/United States
- OT2 OD026552/OD/NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- OT2 OD025337/OD/NIH HHS/United States
- R35 GM122455/GM/NIGMS NIH HHS/United States
- OT2 OD025277/OD/NIH HHS/United States
- MR/S035699/1/MRC_/Medical Research Council/United Kingdom
- T32 GM144273/GM/NIGMS NIH HHS/United States
- OT2 OD026550/OD/NIH HHS/United States
- OT2 OD023205/OD/NIH HHS/United States
- K99 HG012222/HG/NHGRI NIH HHS/United States
- OT2 OD026557/OD/NIH HHS/United States
- R01 MH101244/MH/NIMH NIH HHS/United States
- OT2 OD023206/OD/NIH HHS/United States
- MR/S005021/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Research Materials
