

Epigenetic signature of human immune aging in the GESTALT study
- PMID: 37589453
- PMCID: PMC10506794
- DOI: 10.7554/eLife.86136
Epigenetic signature of human immune aging in the GESTALT study
Abstract
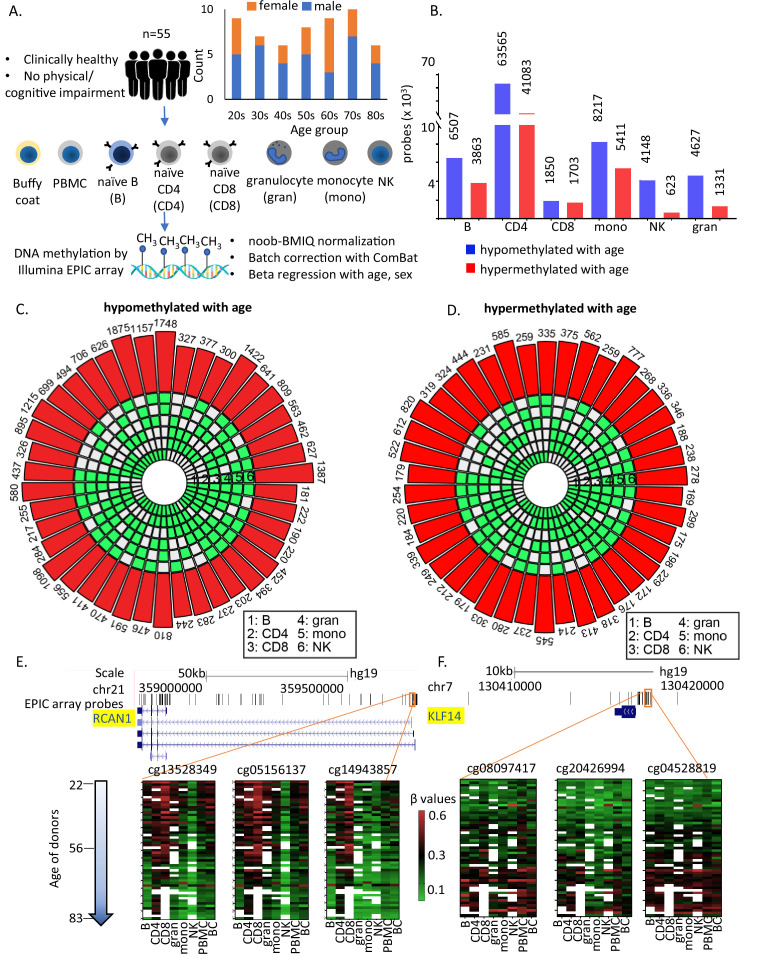
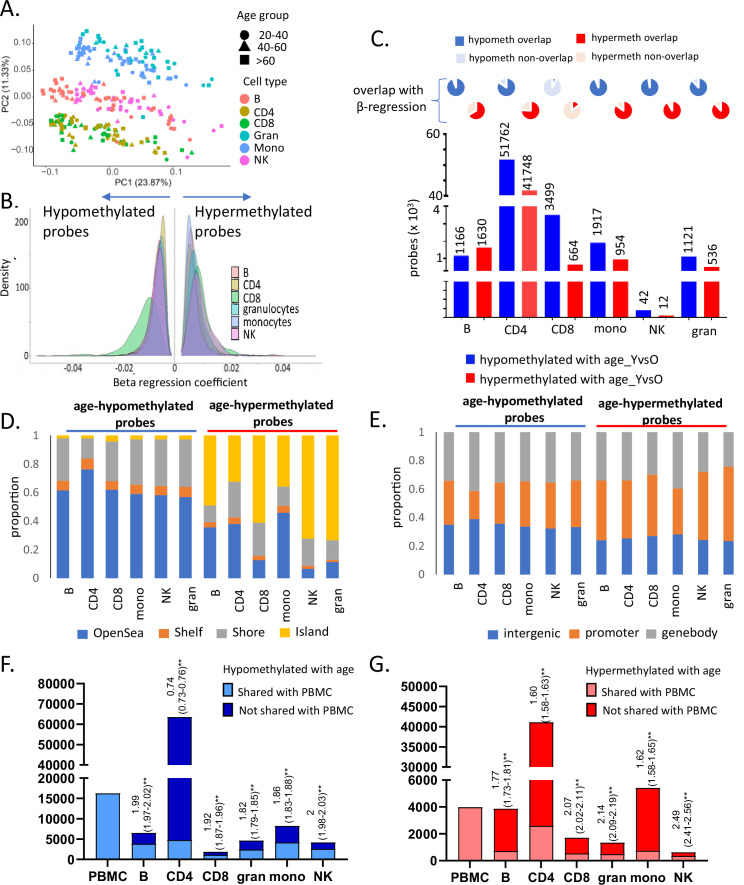
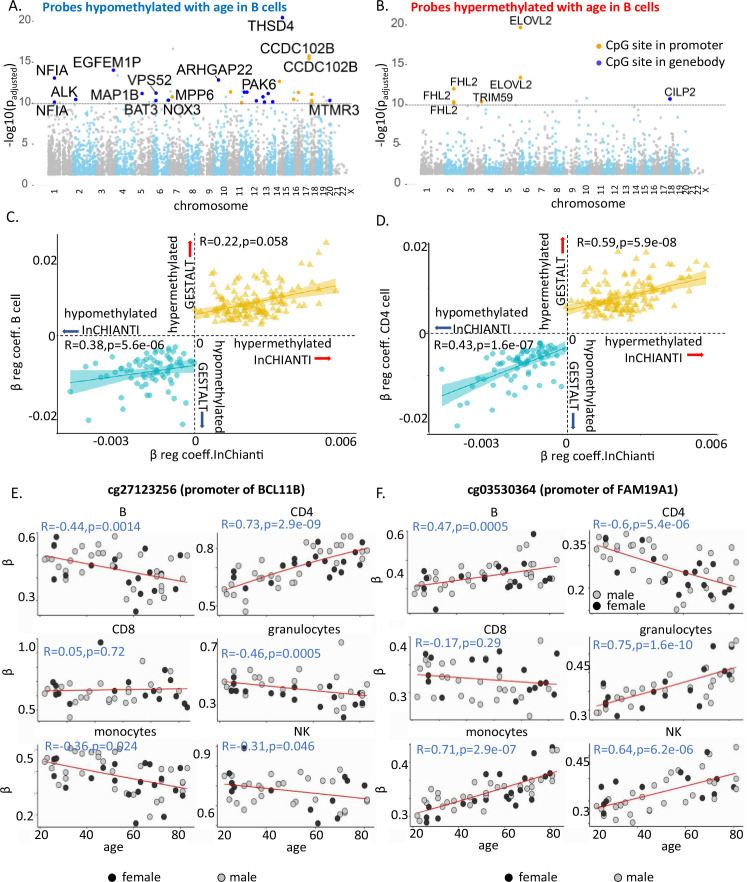
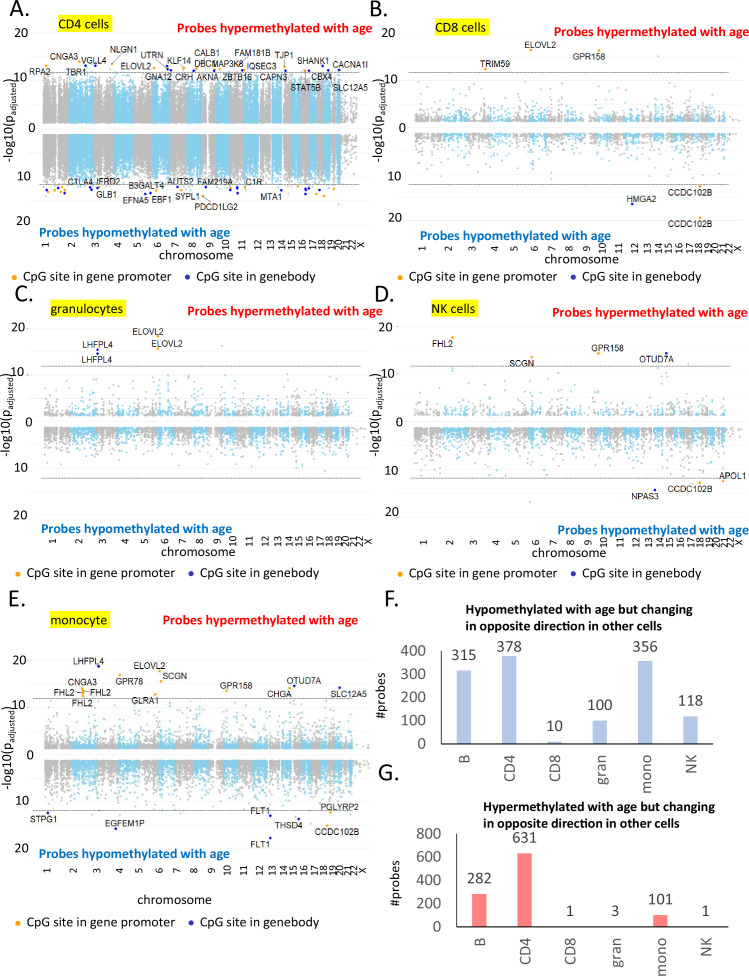
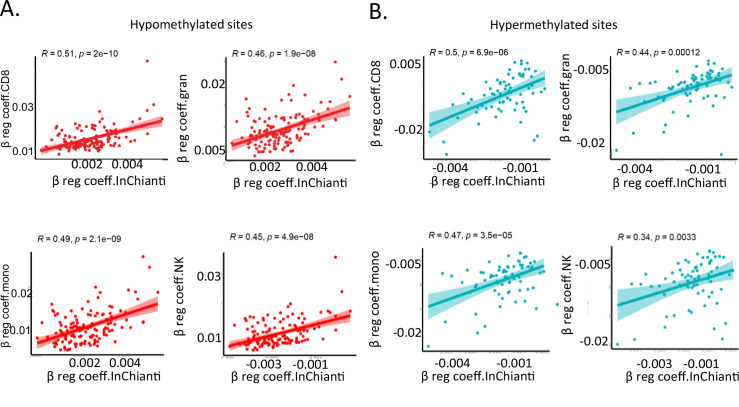
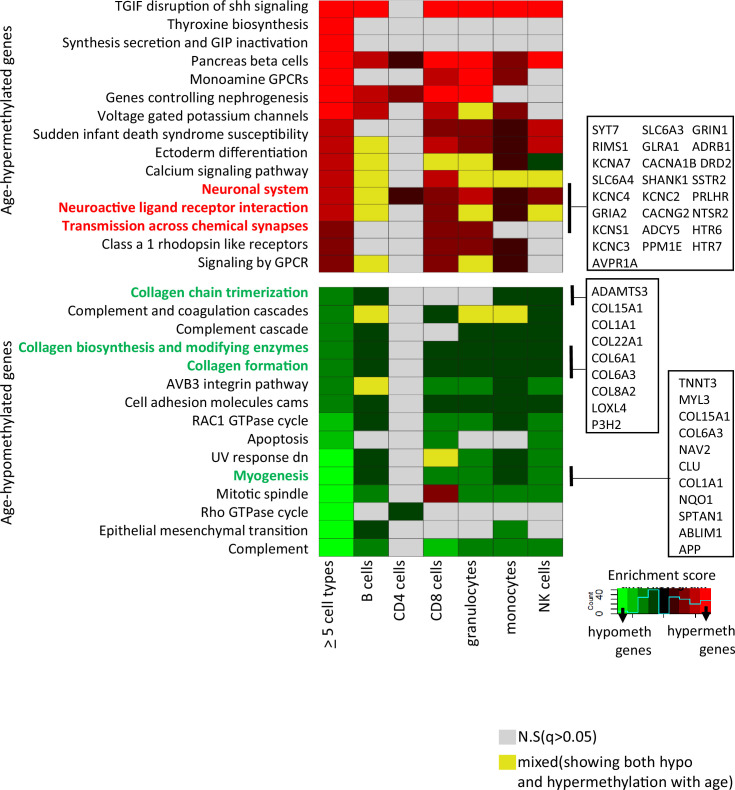
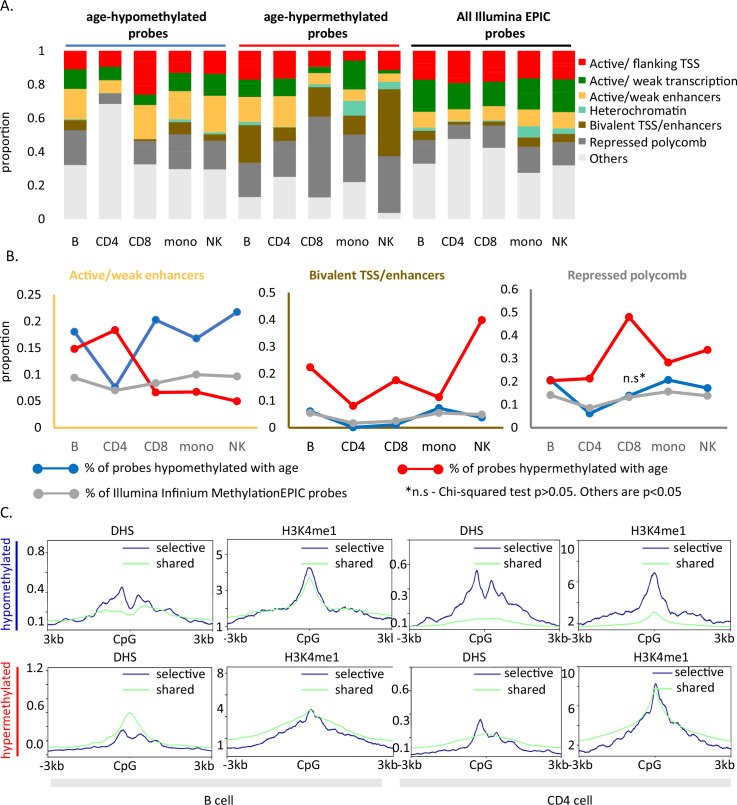
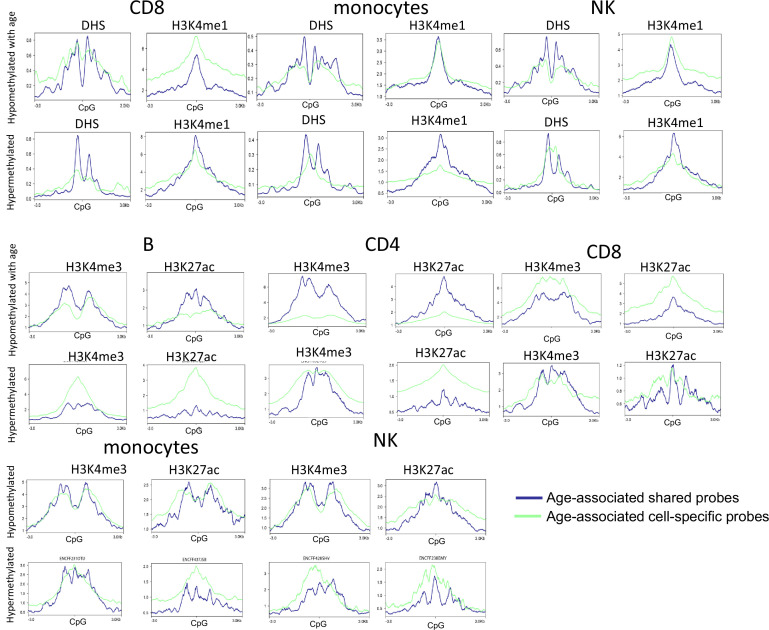
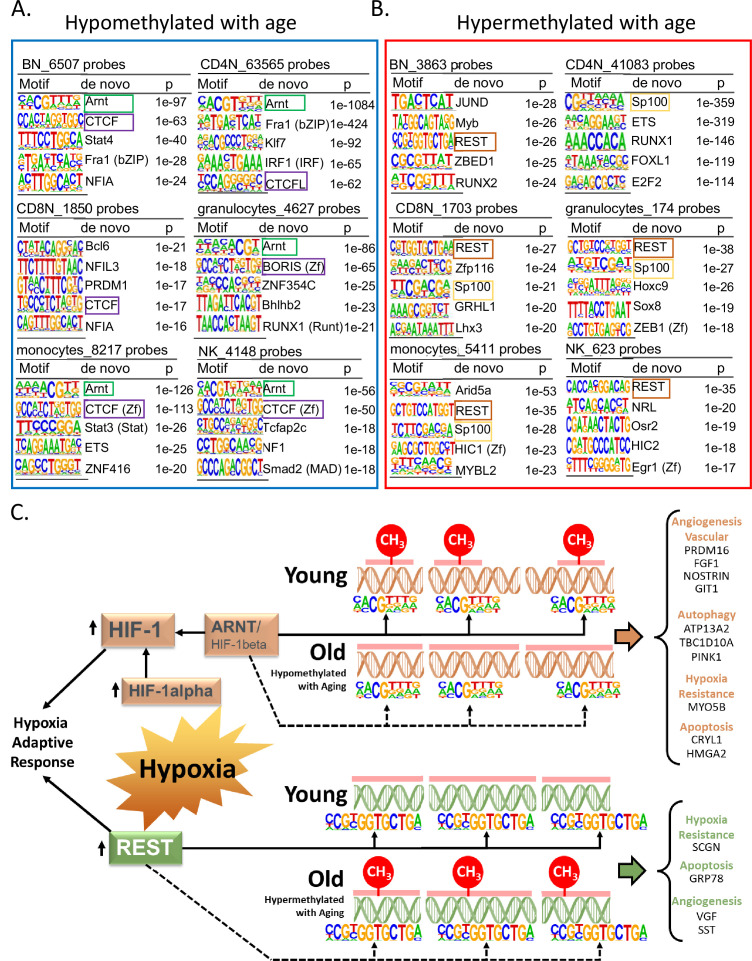
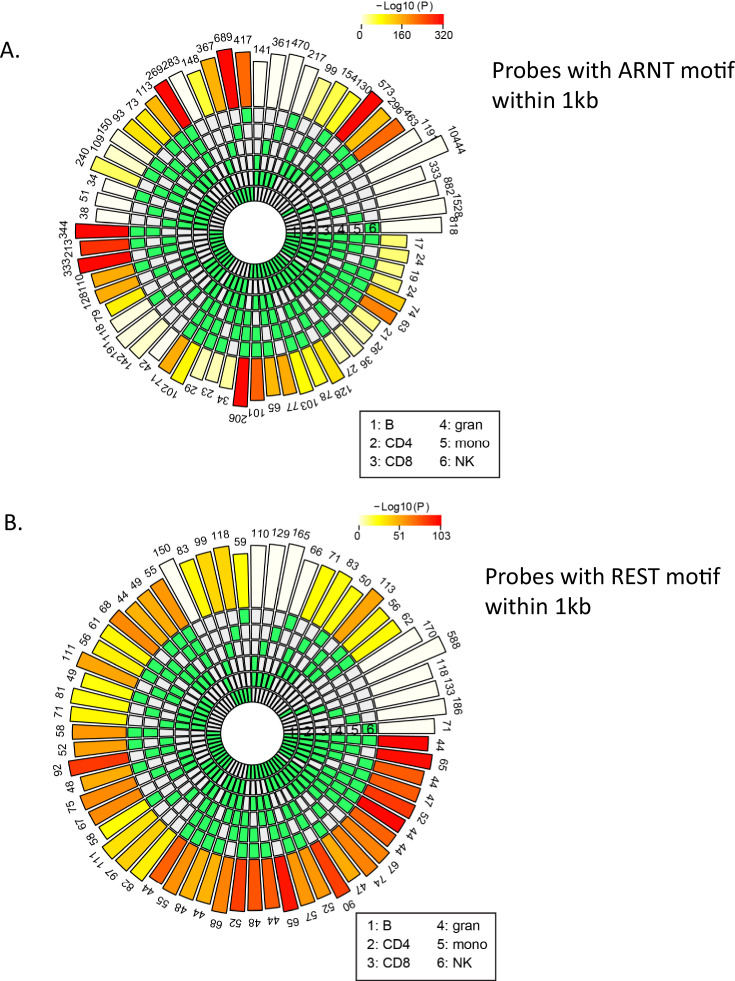
Age-associated DNA methylation in blood cells convey information on health status. However, the mechanisms that drive these changes in circulating cells and their relationships to gene regulation are unknown. We identified age-associated DNA methylation sites in six purified blood-borne immune cell types (naive B, naive CD4+ and CD8+ T cells, granulocytes, monocytes, and NK cells) collected from healthy individuals interspersed over a wide age range. Of the thousands of age-associated sites, only 350 sites were differentially methylated in the same direction in all cell types and validated in an independent longitudinal cohort. Genes close to age-associated hypomethylated sites were enriched for collagen biosynthesis and complement cascade pathways, while genes close to hypermethylated sites mapped to neuronal pathways. In silico analyses showed that in most cell types, the age-associated hypo- and hypermethylated sites were enriched for ARNT (HIF1β) and REST transcription factor (TF) motifs, respectively, which are both master regulators of hypoxia response. To conclude, despite spatial heterogeneity, there is a commonality in the putative regulatory role with respect to TF motifs and histone modifications at and around these sites. These features suggest that DNA methylation changes in healthy aging may be adaptive responses to fluctuations of oxygen availability.
Keywords: DNA methylation; genetics; genomics; healthy aging; human; hypoxia; immune cells; immunology; inflammation.
Conflict of interest statement
RR, PK, JC, DS, CU, DH, MK, SA, AS, AB, JK, AM, TT, JM, LZ, CN, TW, CD, WW, YP, CC, SD, JS, NW, RS, LF No competing interests declared
Figures
Update of
References
-
- Acevedo N, Reinius LE, Vitezic M, Fortino V, Söderhäll C, Honkanen H, Veijola R, Simell O, Toppari J, Ilonen J, Knip M, Scheynius A, Hyöty H, Greco D, Kere J. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clinical Epigenetics. 2015;7:34. doi: 10.1186/s13148-015-0064-6. - DOI - PMC - PubMed
-
- Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, Shin S-Y, Dempster EL, Murray RM, Grundberg E, Hedman AK, Nica A, Small KS, MuTHER Consortium. Dermitzakis ET, McCarthy MI, Mill J, Spector TD, Deloukas P. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLOS Genetics. 2012;8:e1002629. doi: 10.1371/journal.pgen.1002629. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
