

Chemical remodeling of a cellular chaperone to target the active state of mutant KRAS
- PMID: 37590355
- PMCID: PMC10474815
- DOI: 10.1126/science.adg9652
Chemical remodeling of a cellular chaperone to target the active state of mutant KRAS
Abstract
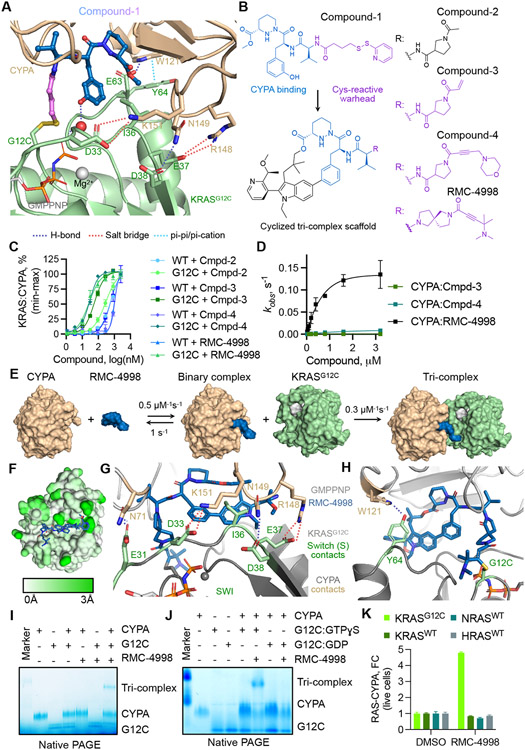
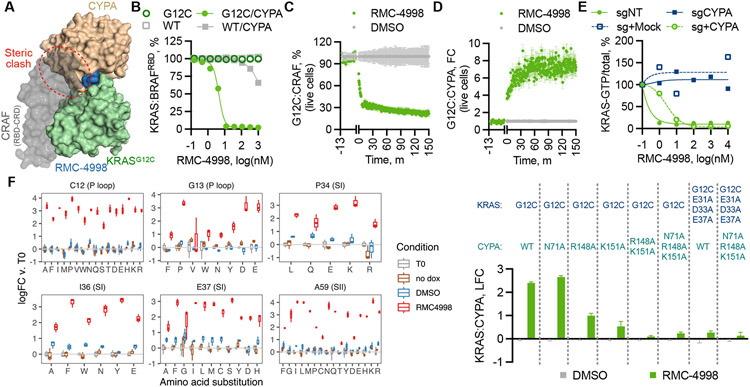
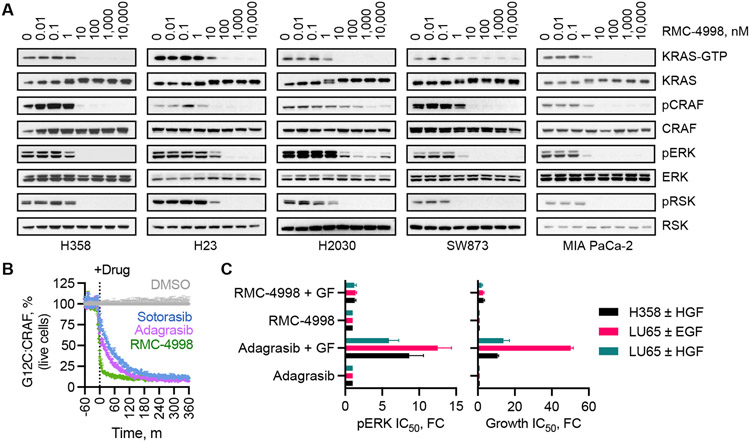
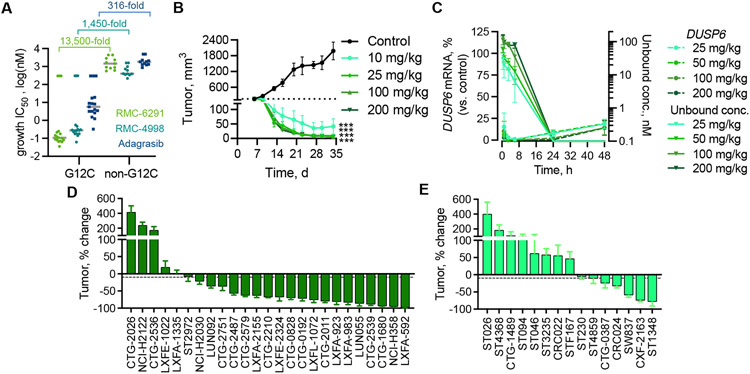
The discovery of small-molecule inhibitors requires suitable binding pockets on protein surfaces. Proteins that lack this feature are considered undruggable and require innovative strategies for therapeutic targeting. KRAS is the most frequently activated oncogene in cancer, and the active state of mutant KRAS is such a recalcitrant target. We designed a natural product-inspired small molecule that remodels the surface of cyclophilin A (CYPA) to create a neomorphic interface with high affinity and selectivity for the active state of KRASG12C (in which glycine-12 is mutated to cysteine). The resulting CYPA:drug:KRASG12C tricomplex inactivated oncogenic signaling and led to tumor regressions in multiple human cancer models. This inhibitory strategy can be used to target additional KRAS mutants and other undruggable cancer drivers. Tricomplex inhibitors that selectively target active KRASG12C or multiple RAS mutants are in clinical trials now (NCT05462717 and NCT05379985).
Figures
References
-
- Malumbres M, Barbacid M, RAS oncogenes: the first 30 years. Nat Rev Cancer 3, 459–465 (2003). - PubMed
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
