

Sequence dependencies and mutation rates of localized mutational processes in cancer
- PMID: 37592287
- PMCID: PMC10436389
- DOI: 10.1186/s13073-023-01217-z
Sequence dependencies and mutation rates of localized mutational processes in cancer
Abstract
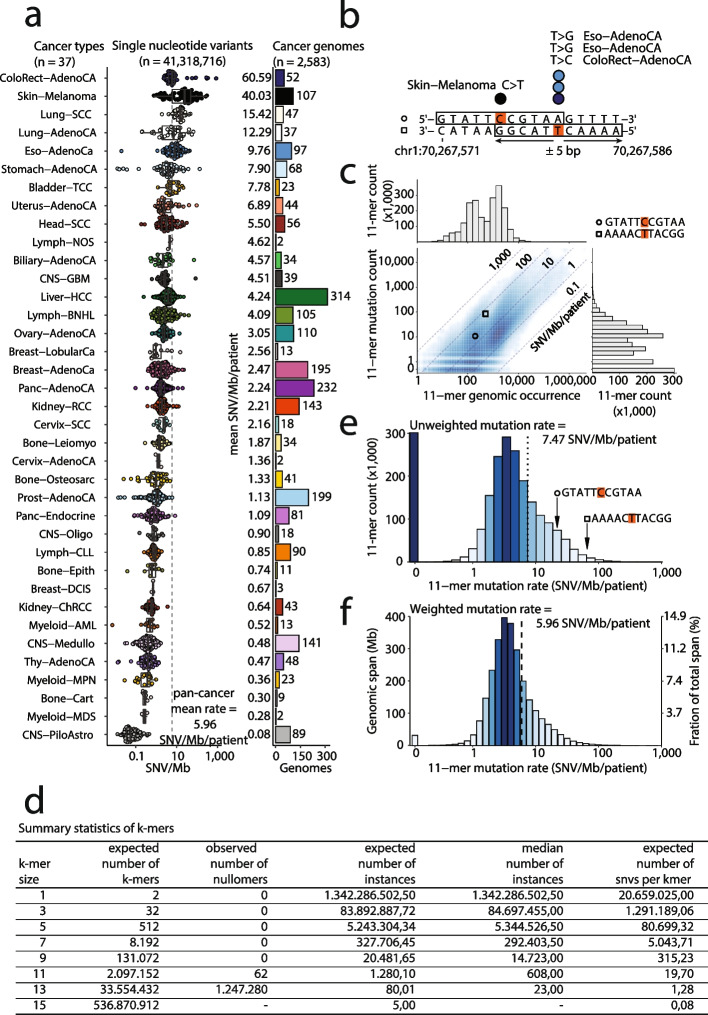
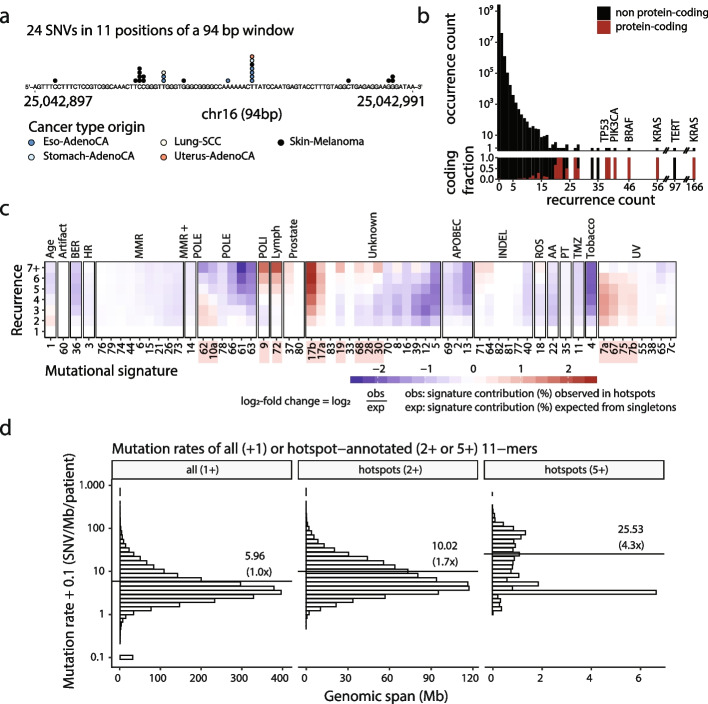
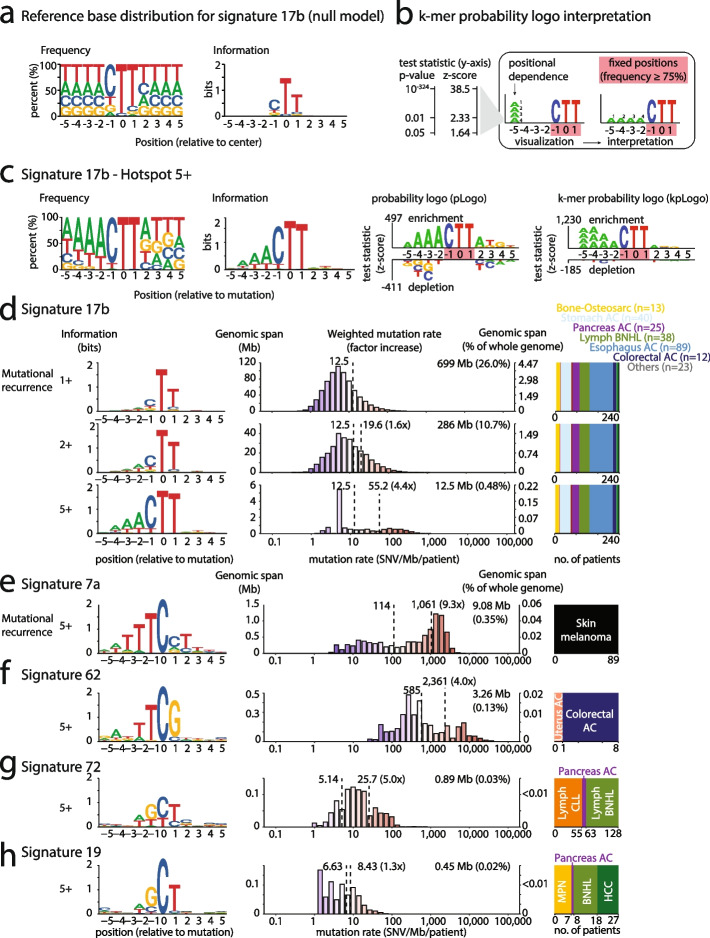
Background: Cancer mutations accumulate through replication errors and DNA damage coupled with incomplete repair. Individual mutational processes often show nucleotide sequence and functional region preferences. As a result, some sequence contexts mutate at much higher rates than others, with additional variation found between functional regions. Mutational hotspots, with recurrent mutations across cancer samples, represent genomic positions with elevated mutation rates, often caused by highly localized mutational processes.
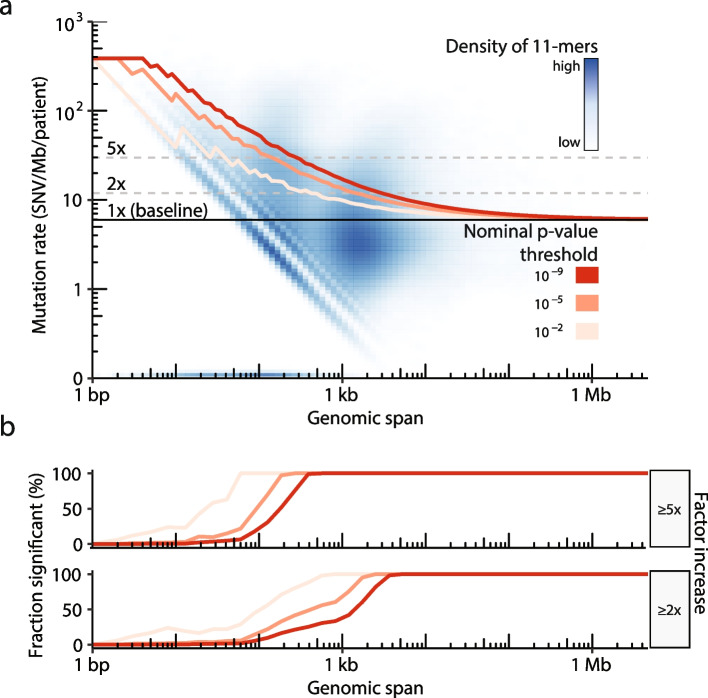
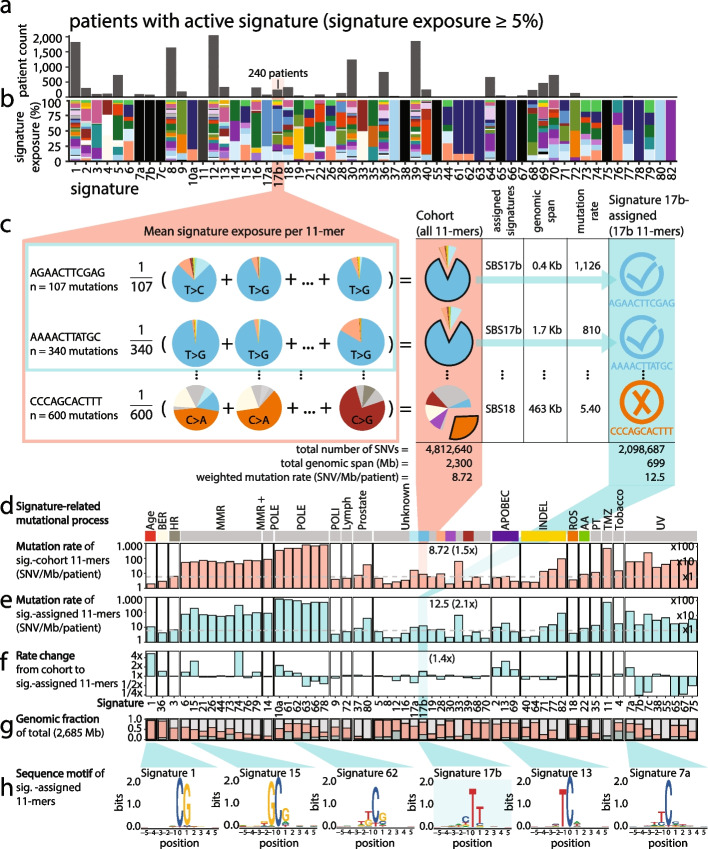
Methods: We count the 11-mer genomic sequences across the genome, and using the PCAWG set of 2583 pan-cancer whole genomes, we associate 11-mers with mutational signatures, hotspots of single nucleotide variants, and specific genomic regions. We evaluate the mutation rates of individual and combined sets of 11-mers and derive mutational sequence motifs.
Results: We show that hotspots generally identify highly mutable sequence contexts. Using these, we show that some mutational signatures are enriched in hotspot sequence contexts, corresponding to well-defined sequence preferences for the underlying localized mutational processes. This includes signature 17b (of unknown etiology) and signatures 62 (POLE deficiency), 7a (UV), and 72 (linked to lymphomas). In some cases, the mutation rate and sequence preference increase further when focusing on certain genomic regions, such as signature 62 in transcribed regions, where the mutation rate is increased up to 9-folds over cancer type and mutational signature average.
Conclusions: We summarize our findings in a catalog of localized mutational processes, their sequence preferences, and their estimated mutation rates.
Keywords: Hotspots; Mutation rate; Mutational processes; Pan-cancer.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
Hotspot mutations delineating diverse mutational signatures and biological utilities across cancer types.BMC Genomics. 2016 Jun 23;17 Suppl 2(Suppl 2):394. doi: 10.1186/s12864-016-2727-x. BMC Genomics. 2016. PMID: 27356755 Free PMC article.
-
Hotspot propensity across mutational processes.Mol Syst Biol. 2024 Jan;20(1):6-27. doi: 10.1038/s44320-023-00001-w. Epub 2023 Dec 20. Mol Syst Biol. 2024. PMID: 38177930 Free PMC article.
-
Regional mutational signature activities in cancer genomes.PLoS Comput Biol. 2022 Dec 5;18(12):e1010733. doi: 10.1371/journal.pcbi.1010733. eCollection 2022 Dec. PLoS Comput Biol. 2022. PMID: 36469539 Free PMC article.
-
Mutational signatures and mutable motifs in cancer genomes.Brief Bioinform. 2018 Nov 27;19(6):1085-1101. doi: 10.1093/bib/bbx049. Brief Bioinform. 2018. PMID: 28498882 Free PMC article. Review.
-
Analysis of mutational signatures in C. elegans: Implications for cancer genome analysis.DNA Repair (Amst). 2020 Nov;95:102957. doi: 10.1016/j.dnarep.2020.102957. Epub 2020 Aug 28. DNA Repair (Amst). 2020. PMID: 32980770 Review.
Cited by
-
MAFcounter: An efficient tool for counting the occurrences of k-mers in MAF files.ArXiv [Preprint]. 2024 Nov 29:arXiv:2411.19427v1. ArXiv. 2024. Update in: BMC Bioinformatics. 2025 May 30;26(1):142. doi: 10.1186/s12859-025-06172-7. PMID: 39650609 Free PMC article. Updated. Preprint.
-
MAFcounter: an efficient tool for counting the occurrences of k-mers in MAF files.BMC Bioinformatics. 2025 May 30;26(1):142. doi: 10.1186/s12859-025-06172-7. BMC Bioinformatics. 2025. PMID: 40448014 Free PMC article.
-
A prognostic model for laryngeal squamous cell carcinoma based on the mitochondrial metabolism-related genes.Transl Cancer Res. 2025 Feb 28;14(2):966-979. doi: 10.21037/tcr-24-1436. Epub 2025 Feb 18. Transl Cancer Res. 2025. PMID: 40104737 Free PMC article.
-
Target-Enhanced Whole-Genome Sequencing Shows Clinical Validity Equivalent to Commercially Available Targeted Oncology Panel.Cancer Res Treat. 2025 Apr;57(2):350-361. doi: 10.4143/crt.2024.114. Epub 2024 Sep 19. Cancer Res Treat. 2025. PMID: 39300929 Free PMC article.
-
kmerDB: A database encompassing the set of genomic and proteomic sequence information for each species.Comput Struct Biotechnol J. 2024 Apr 21;23:1919-1928. doi: 10.1016/j.csbj.2024.04.050. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38711760 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
