

Genome-wide transcription factor-binding maps reveal cell-specific changes in the regulatory architecture of human HSPCs
- PMID: 37595278
- PMCID: PMC10651876
- DOI: 10.1182/blood.2023021120
Genome-wide transcription factor-binding maps reveal cell-specific changes in the regulatory architecture of human HSPCs
Abstract
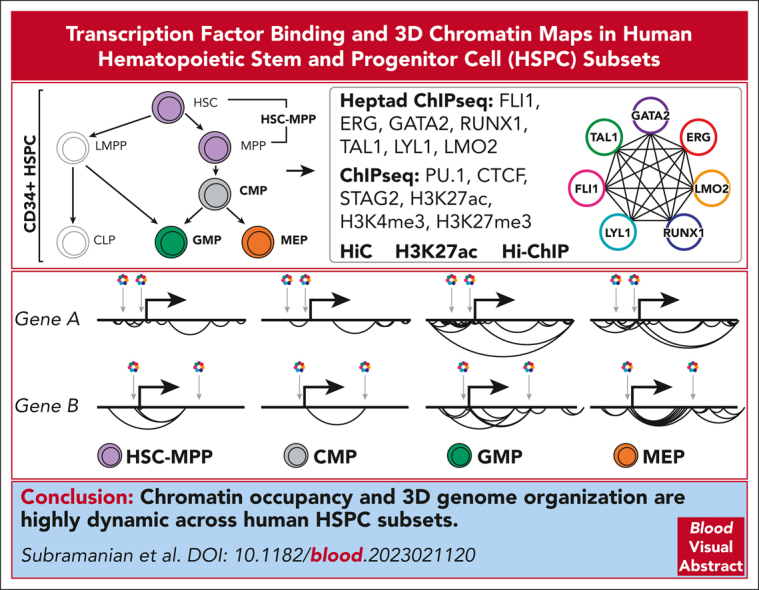
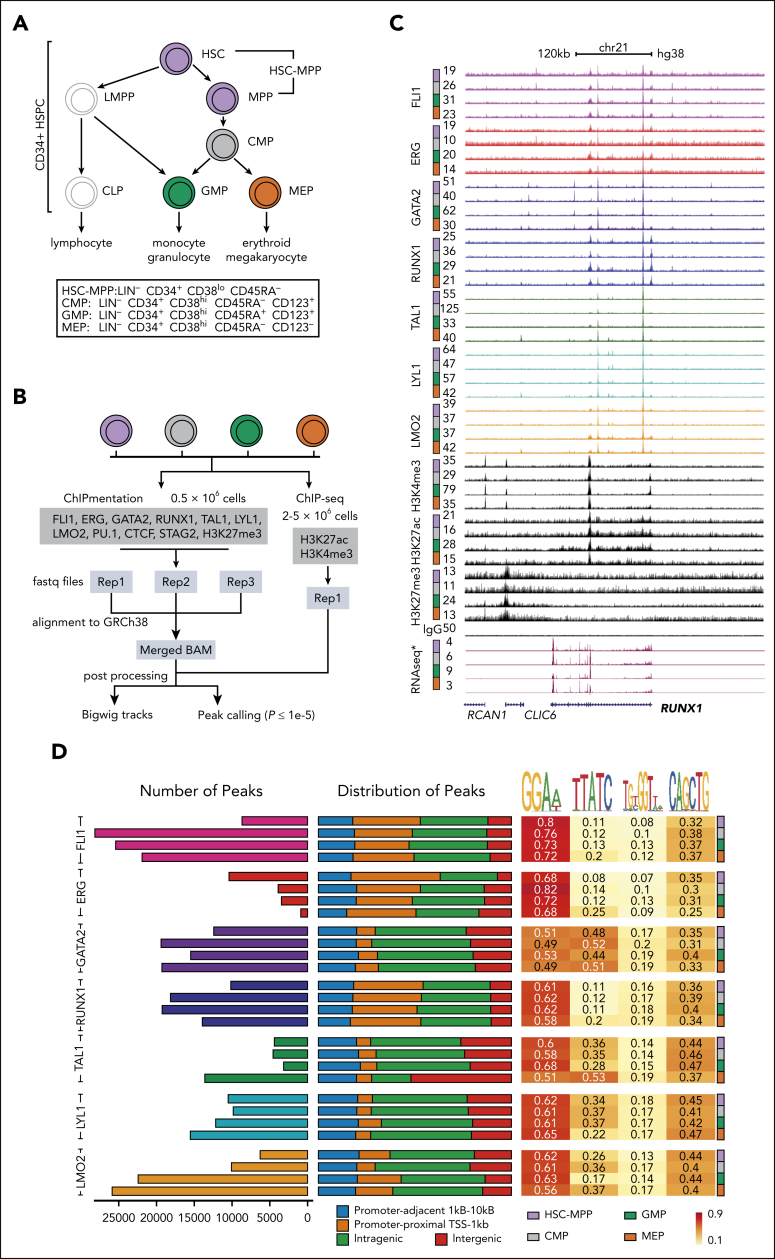
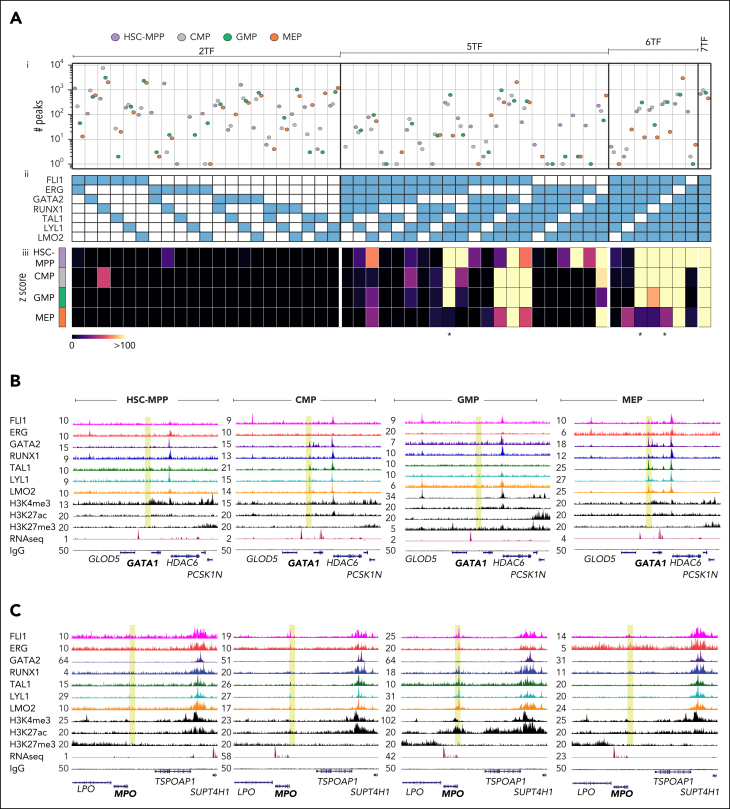
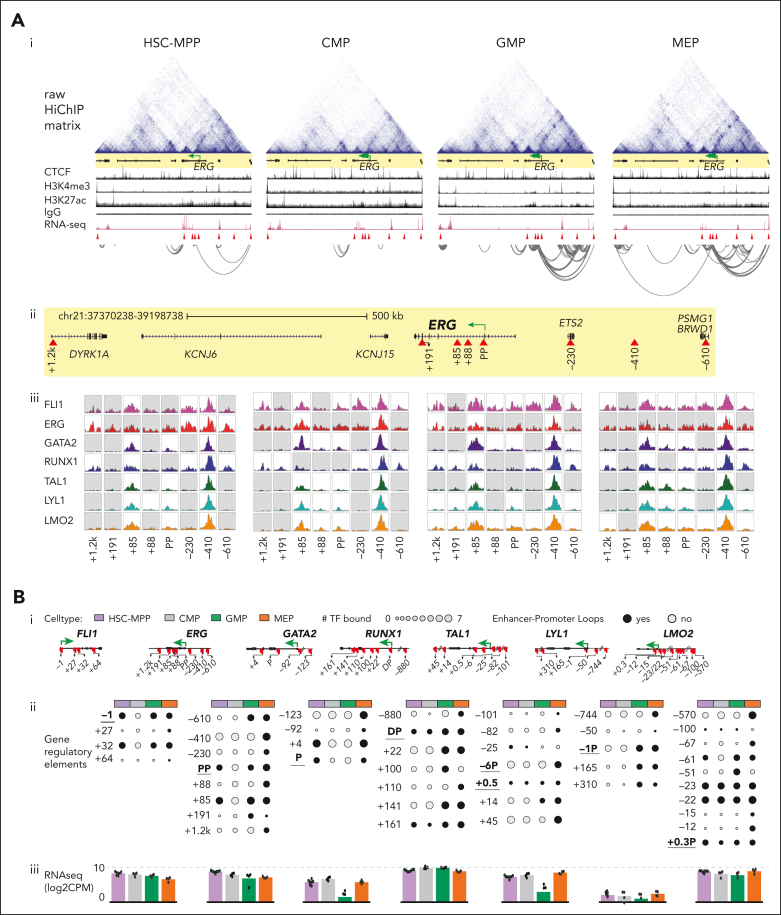
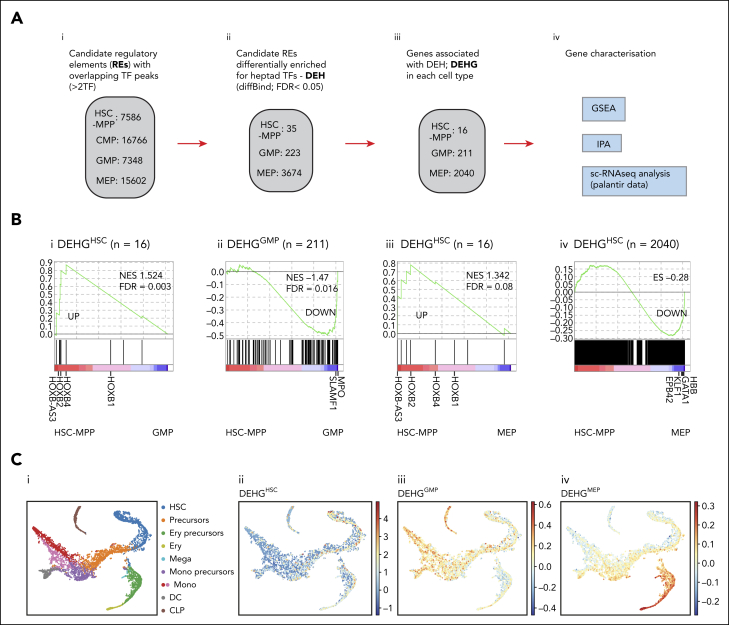
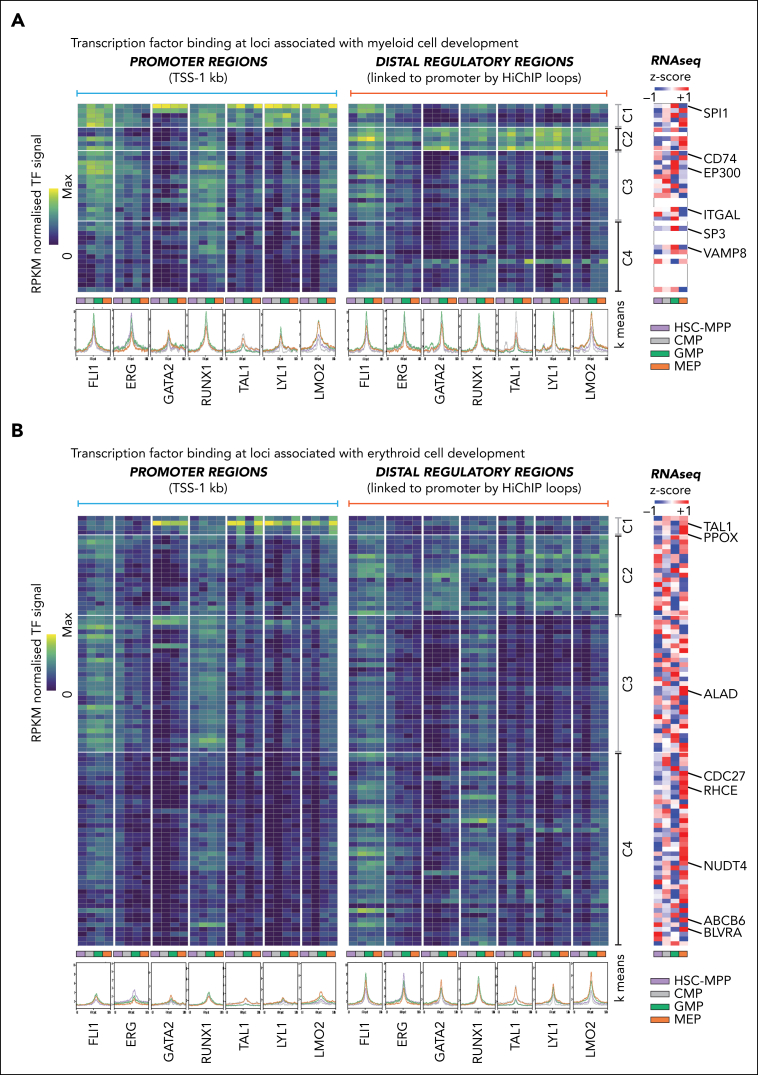
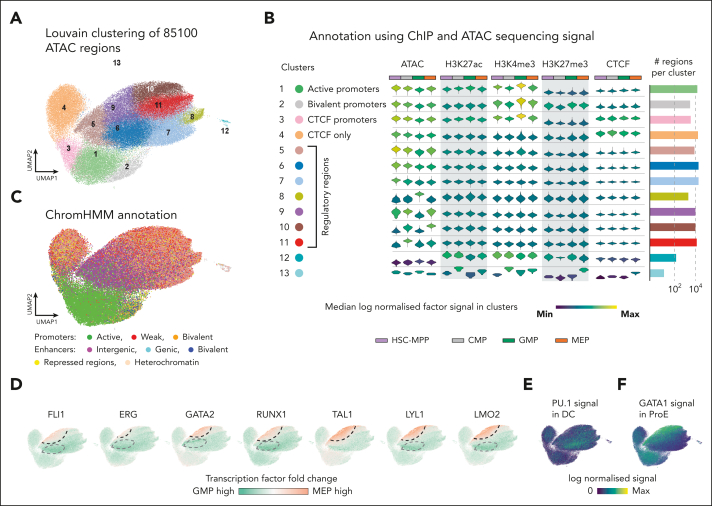
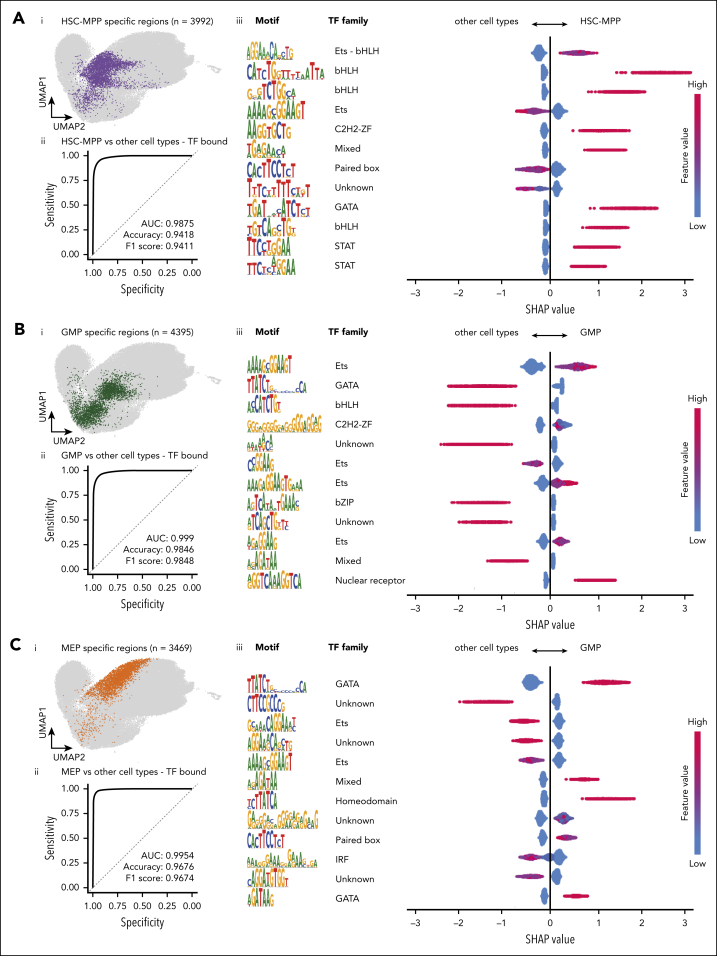
Hematopoietic stem and progenitor cells (HSPCs) rely on a complex interplay among transcription factors (TFs) to regulate differentiation into mature blood cells. A heptad of TFs (FLI1, ERG, GATA2, RUNX1, TAL1, LYL1, LMO2) bind regulatory elements in bulk CD34+ HSPCs. However, whether specific heptad-TF combinations have distinct roles in regulating hematopoietic differentiation remains unknown. We mapped genome-wide chromatin contacts (HiC, H3K27ac, HiChIP), chromatin modifications (H3K4me3, H3K27ac, H3K27me3) and 10 TF binding profiles (heptad, PU.1, CTCF, STAG2) in HSPC subsets (stem/multipotent progenitors plus common myeloid, granulocyte macrophage, and megakaryocyte erythrocyte progenitors) and found TF occupancy and enhancer-promoter interactions varied significantly across cell types and were associated with cell-type-specific gene expression. Distinct regulatory elements were enriched with specific heptad-TF combinations, including stem-cell-specific elements with ERG, and myeloid- and erythroid-specific elements with combinations of FLI1, RUNX1, GATA2, TAL1, LYL1, and LMO2. Furthermore, heptad-occupied regions in HSPCs were subsequently bound by lineage-defining TFs, including PU.1 and GATA1, suggesting that heptad factors may prime regulatory elements for use in mature cell types. We also found that enhancers with cell-type-specific heptad occupancy shared a common grammar with respect to TF binding motifs, suggesting that combinatorial binding of TF complexes was at least partially regulated by features encoded in DNA sequence motifs. Taken together, this study comprehensively characterizes the gene regulatory landscape in rare subpopulations of human HSPCs. The accompanying data sets should serve as a valuable resource for understanding adult hematopoiesis and a framework for analyzing aberrant regulatory networks in leukemic cells.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
All wired up: heptads in hematopoiesis.Blood. 2023 Oct 26;142(17):1407-1409. doi: 10.1182/blood.2023022014. Blood. 2023. PMID: 37883110 No abstract available.
References
-
- Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–136. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
