

Etiology of lipid-laden macrophages in the lung
- PMID: 37595492
- PMCID: PMC10734282
- DOI: 10.1016/j.intimp.2023.110719
Etiology of lipid-laden macrophages in the lung
Abstract
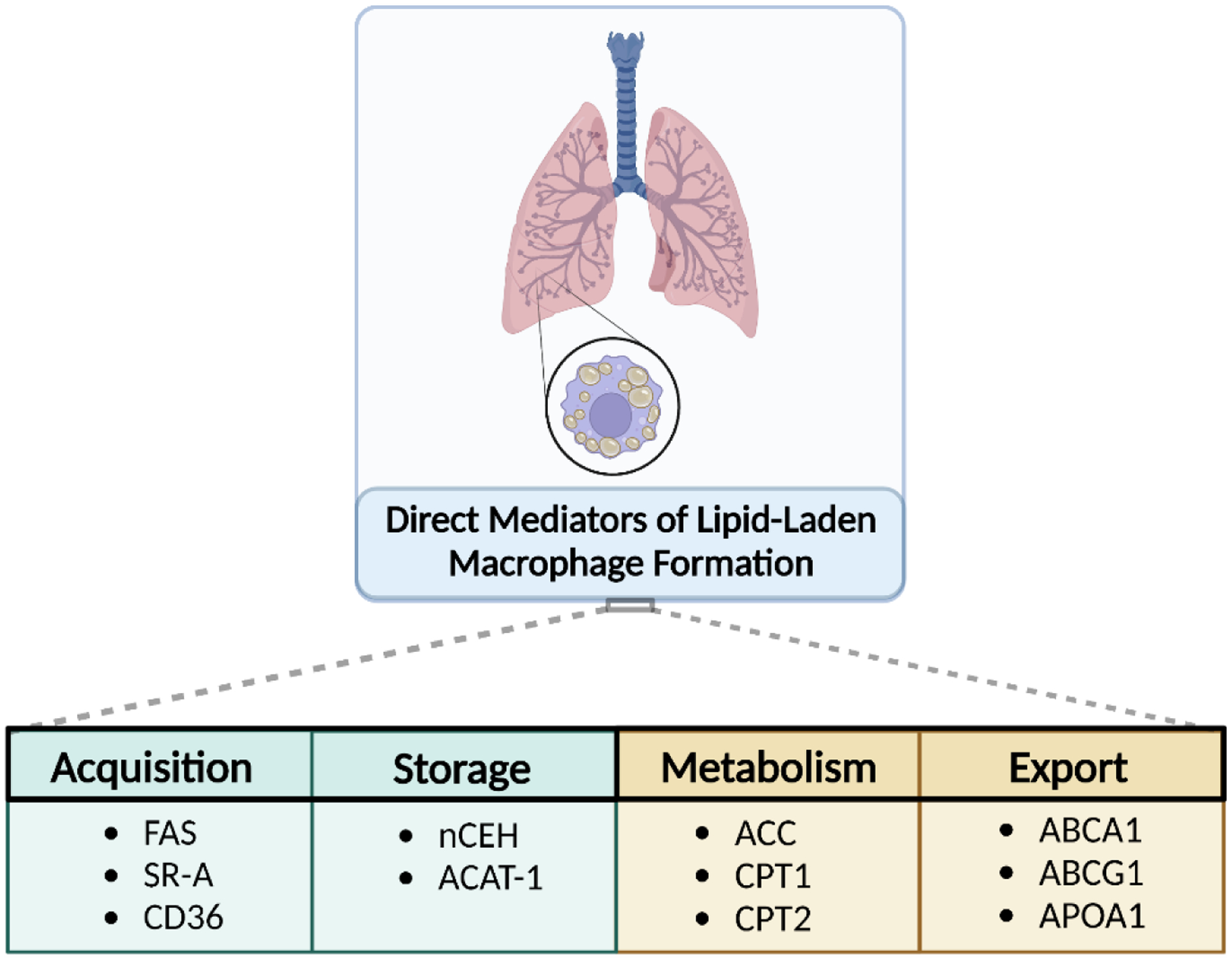
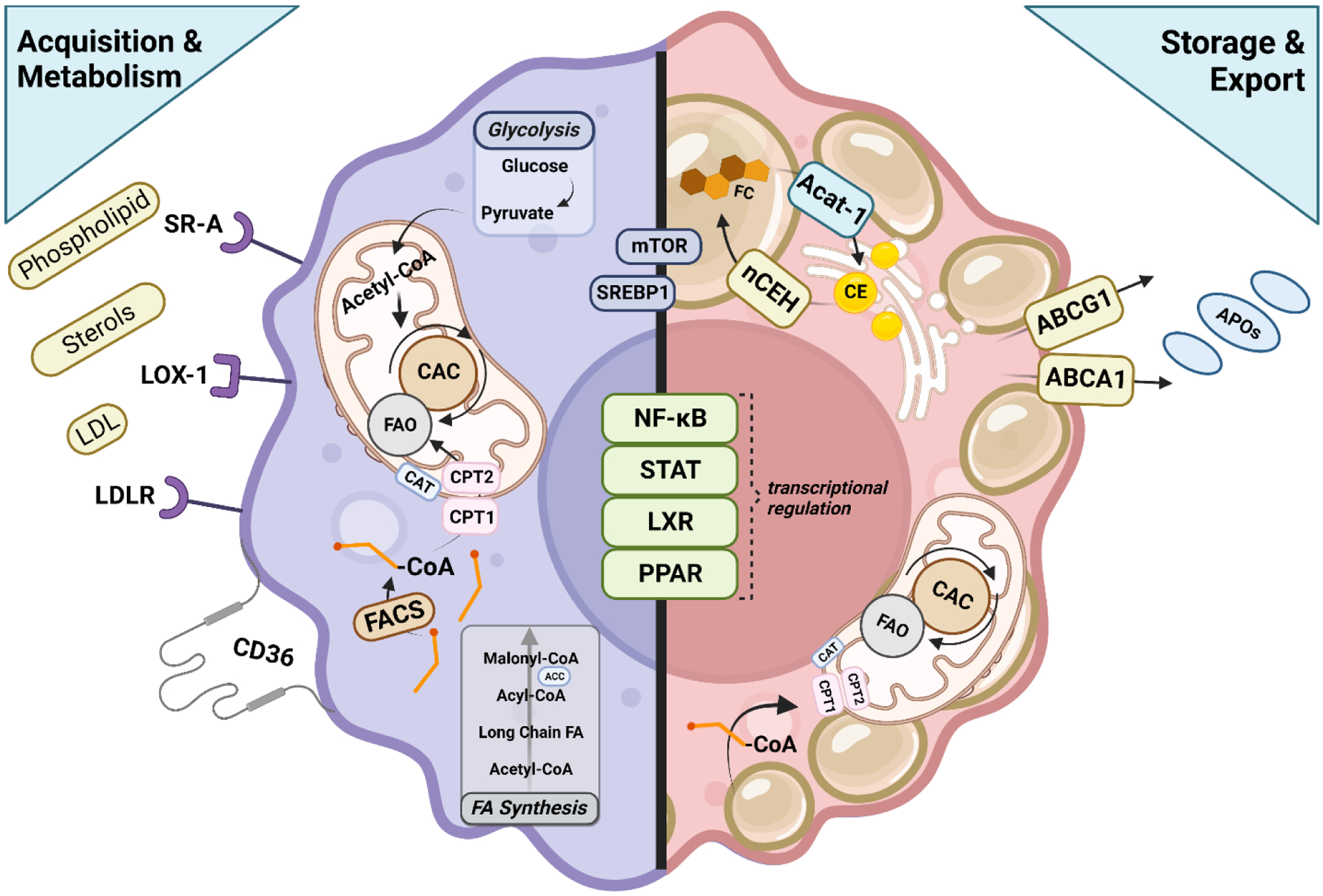
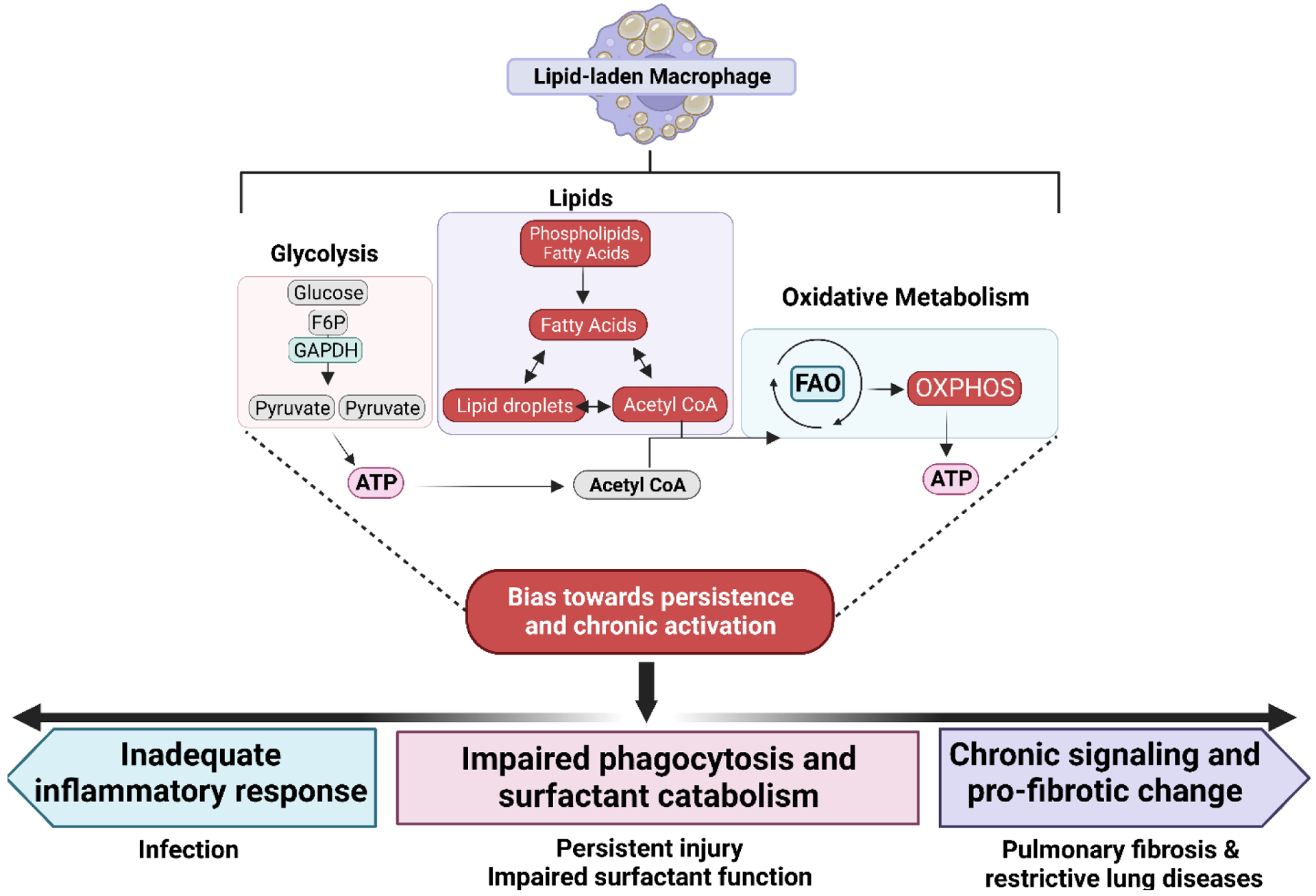
Uniquely positioned as sentinel cells constantly exposed to the environment, pulmonary macrophages are vital for the maintenance of the lung lining. These cells are responsible for the clearance of xenobiotics, pathogen detection and clearance, and homeostatic functions such as surfactant recycling. Among the spectrum of phenotypes that may be expressed by macrophages in the lung, the pulmonary lipid-laden phenotype is less commonly studied in comparison to its circulatory counterpart, the atherosclerotic lesion-associated foam cell, or the acutely activated inflammatory macrophage. Herein, we propose that lipid-laden macrophage formation in the lung is governed by lipid acquisition, storage, metabolism, and export processes. The cellular balance of these four processes is critical to the maintenance of homeostasis and the prevention of aberrant signaling that may contribute to lung pathologies. This review aims to examine mechanisms and signaling pathways that are involved in lipid-laden macrophage formation and the potential consequences of this phenotype in the lung.
Keywords: Foam cell; Immunometabolism; Inflammation; Lipid; Lung; Macrophage.
Copyright © 2023 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
