

Systematic evaluation of genome sequencing for the diagnostic assessment of autism spectrum disorder and fetal structural anomalies
- PMID: 37595579
- PMCID: PMC10502737
- DOI: 10.1016/j.ajhg.2023.07.010
Systematic evaluation of genome sequencing for the diagnostic assessment of autism spectrum disorder and fetal structural anomalies
Abstract
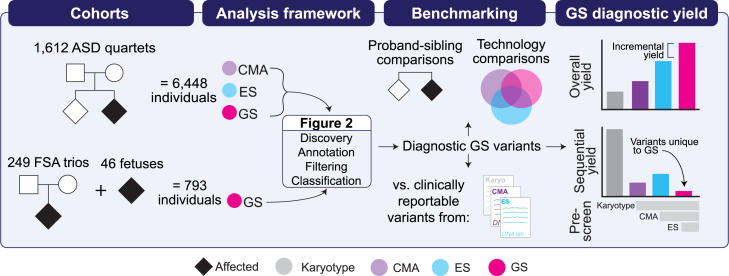
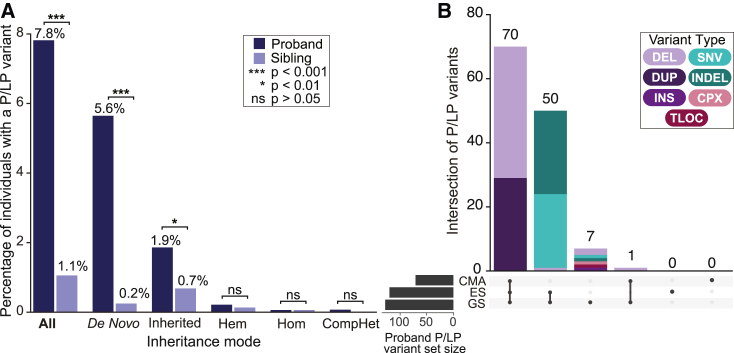
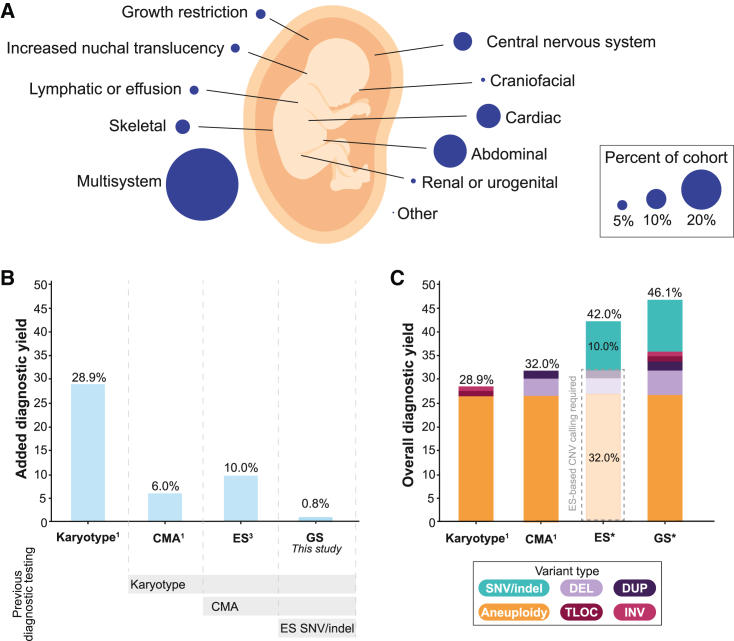
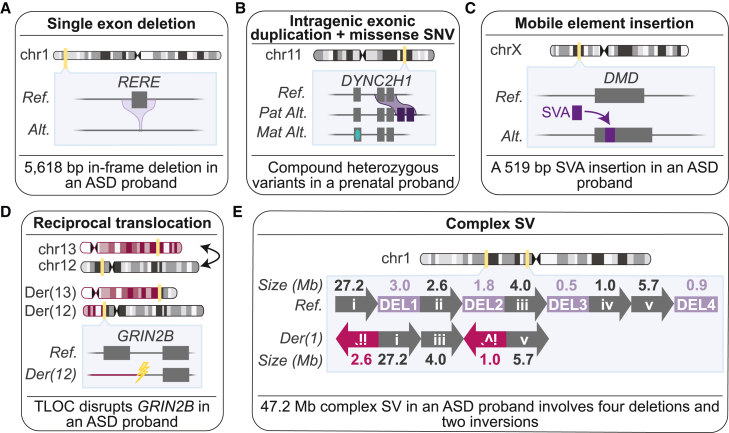
Short-read genome sequencing (GS) holds the promise of becoming the primary diagnostic approach for the assessment of autism spectrum disorder (ASD) and fetal structural anomalies (FSAs). However, few studies have comprehensively evaluated its performance against current standard-of-care diagnostic tests: karyotype, chromosomal microarray (CMA), and exome sequencing (ES). To assess the clinical utility of GS, we compared its diagnostic yield against these three tests in 1,612 quartet families including an individual with ASD and in 295 prenatal families. Our GS analytic framework identified a diagnostic variant in 7.8% of ASD probands, almost 2-fold more than CMA (4.3%) and 3-fold more than ES (2.7%). However, when we systematically captured copy-number variants (CNVs) from the exome data, the diagnostic yield of ES (7.4%) was brought much closer to, but did not surpass, GS. Similarly, we estimated that GS could achieve an overall diagnostic yield of 46.1% in unselected FSAs, representing a 17.2% increased yield over karyotype, 14.1% over CMA, and 4.1% over ES with CNV calling or 36.1% increase without CNV discovery. Overall, GS provided an added diagnostic yield of 0.4% and 0.8% beyond the combination of all three standard-of-care tests in ASD and FSAs, respectively. This corresponded to nine GS unique diagnostic variants, including sequence variants in exons not captured by ES, structural variants (SVs) inaccessible to existing standard-of-care tests, and SVs where the resolution of GS changed variant classification. Overall, this large-scale evaluation demonstrated that GS significantly outperforms each individual standard-of-care test while also outperforming the combination of all three tests, thus warranting consideration as the first-tier diagnostic approach for the assessment of ASD and FSAs.
Keywords: genome sequencing, karyotype, microarray, exome sequencing, structural variant, autism spectrum disorder, structural anomaly, prenatal, first-tier, diagnostic.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.E.T. and H.R. receive research funding from Microsoft Inc and/or research reagents from Illumina Inc. M.E.T. also received research funding from Levo Therapeutics and research reagents from Ionis Therapeutics for unrelated research projects.
Figures

References
-
- Petrovski S., Aggarwal V., Giordano J.L., Stosic M., Wou K., Bier L., Spiegel E., Brennan K., Stong N., Jobanputra V., et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758–767. - PubMed
