

Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response
- PMID: 37596273
- PMCID: PMC10439131
- DOI: 10.1038/s41467-023-40271-4
Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response
Abstract
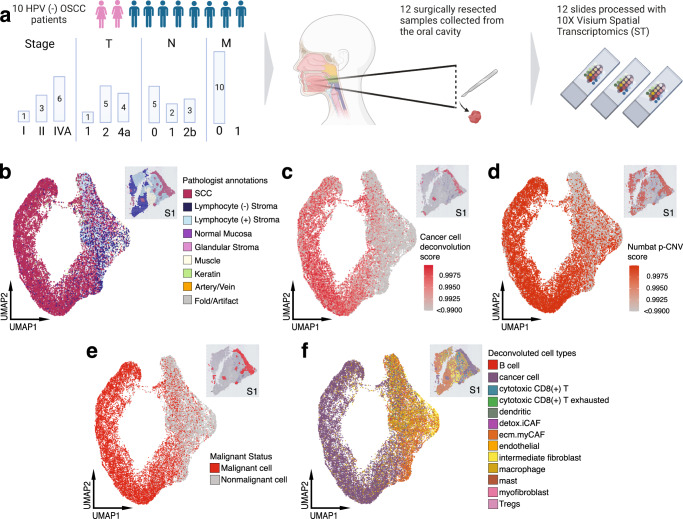
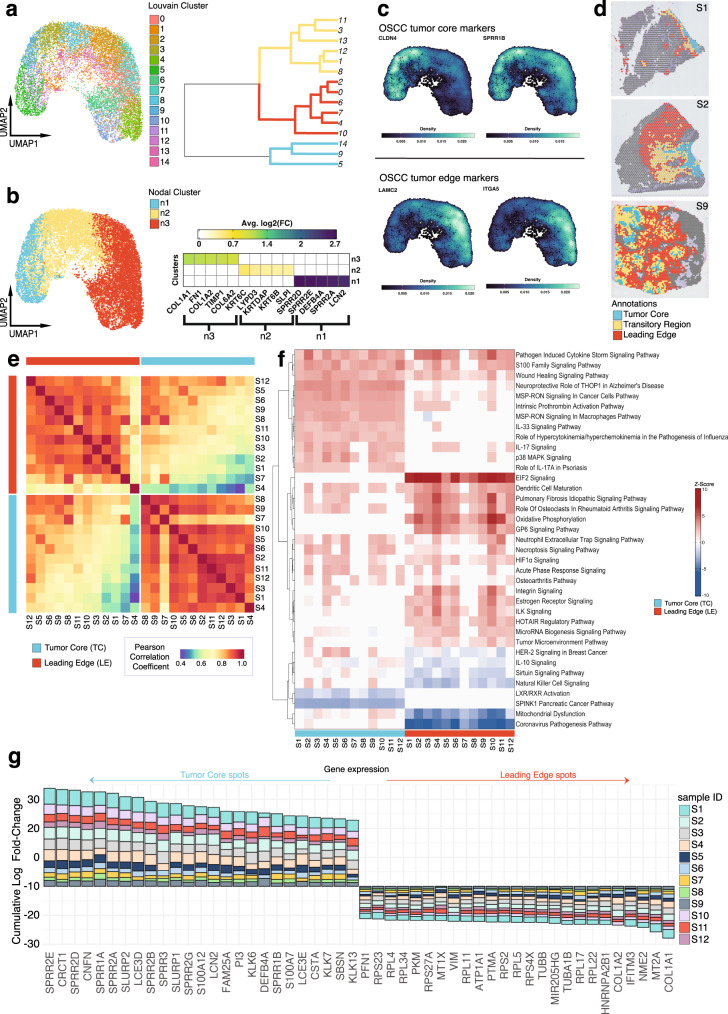
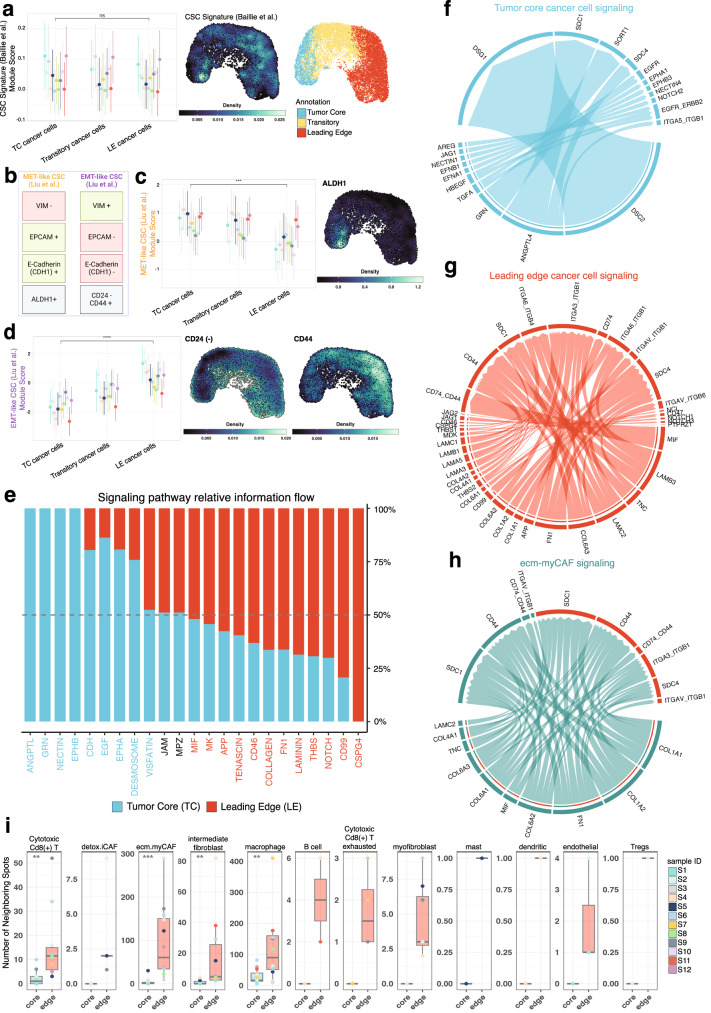
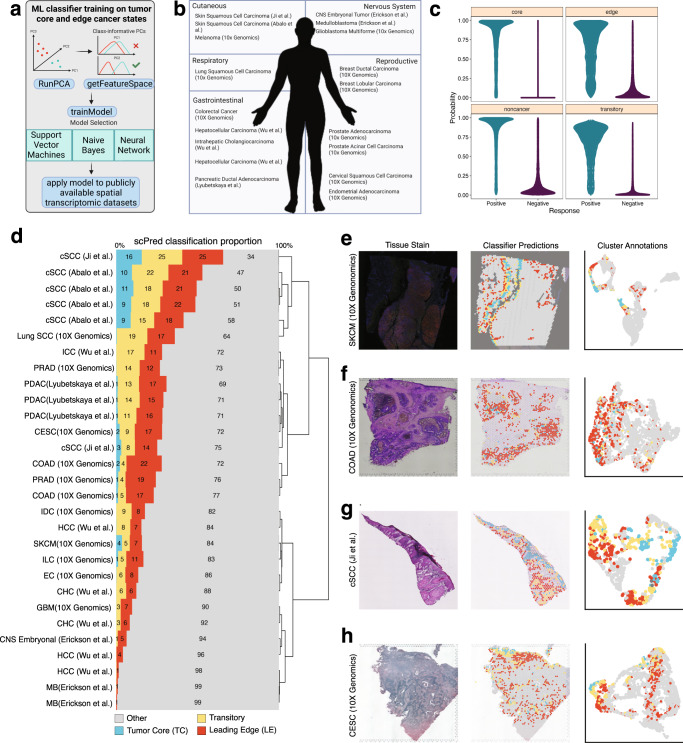
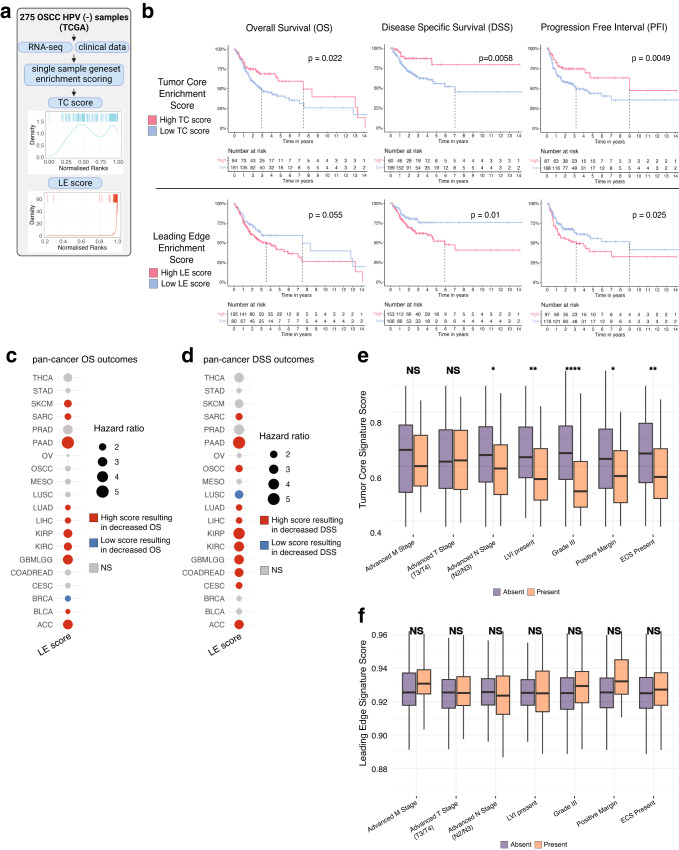
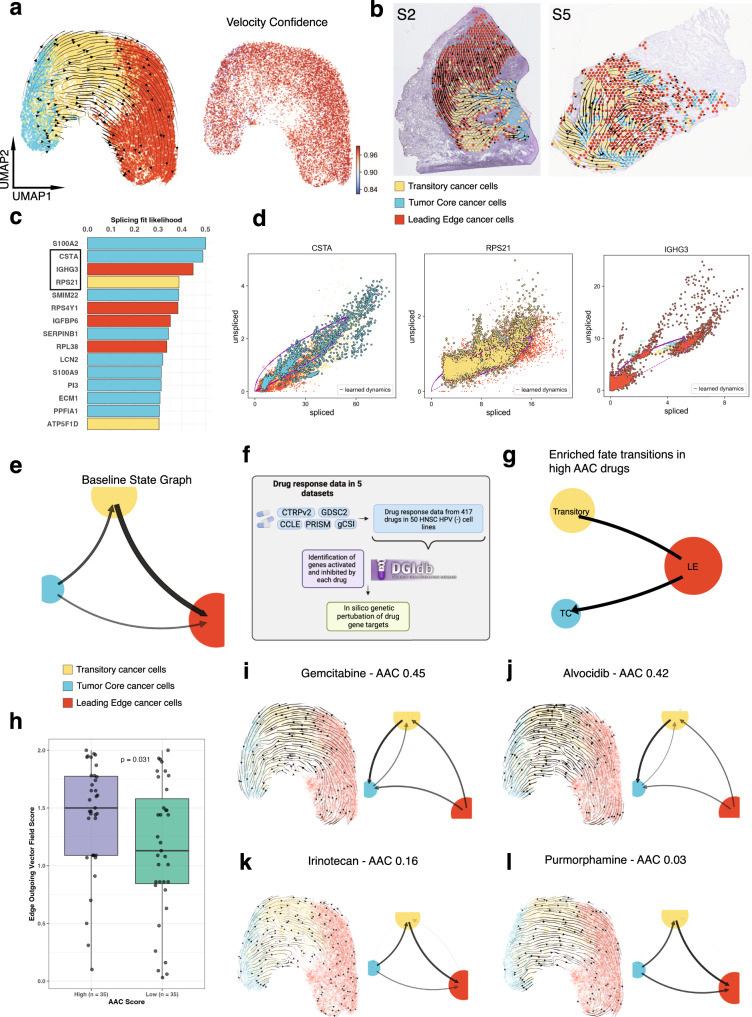
The spatial organization of the tumor microenvironment has a profound impact on biology and therapy response. Here, we perform an integrative single-cell and spatial transcriptomic analysis on HPV-negative oral squamous cell carcinoma (OSCC) to comprehensively characterize malignant cells in tumor core (TC) and leading edge (LE) transcriptional architectures. We show that the TC and LE are characterized by unique transcriptional profiles, neighboring cellular compositions, and ligand-receptor interactions. We demonstrate that the gene expression profile associated with the LE is conserved across different cancers while the TC is tissue specific, highlighting common mechanisms underlying tumor progression and invasion. Additionally, we find our LE gene signature is associated with worse clinical outcomes while TC gene signature is associated with improved prognosis across multiple cancer types. Finally, using an in silico modeling approach, we describe spatially-regulated patterns of cell development in OSCC that are predictably associated with drug response. Our work provides pan-cancer insights into TC and LE biology and interactive spatial atlases ( http://www.pboselab.ca/spatial_OSCC/ ; http://www.pboselab.ca/dynamo_OSCC/ ) that can be foundational for developing novel targeted therapies.
© 2023. Springer Nature Limited.
Conflict of interest statement
R.A. was a bioinformatics consultant at Phenomic AI. R.K.A was previously a Venture Fellow at Flagship Pioneering. R.K.A has served as a Technical Consultant for the Bill and Melinda Gates Foundation Strategic Investment Fund and was a former Senior Policy Advisor at Health Canada. R.K.A also holds a minority stake in Alethea Medical. P.B. is the co-founder and Vice President of Management and Planning at OncoHelix, Inc. All other study authors declare no competing interests.
Figures
References
-
- Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.71, 209–249 (2021). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
