

Role of the DEAD-box RNA helicase DDX5 (p68) in cancer DNA repair, immune suppression, cancer metabolic control, virus infection promotion, and human microbiome (microbiota) negative influence
- PMID: 37596619
- PMCID: PMC10439624
- DOI: 10.1186/s13046-023-02787-x
Role of the DEAD-box RNA helicase DDX5 (p68) in cancer DNA repair, immune suppression, cancer metabolic control, virus infection promotion, and human microbiome (microbiota) negative influence
Abstract
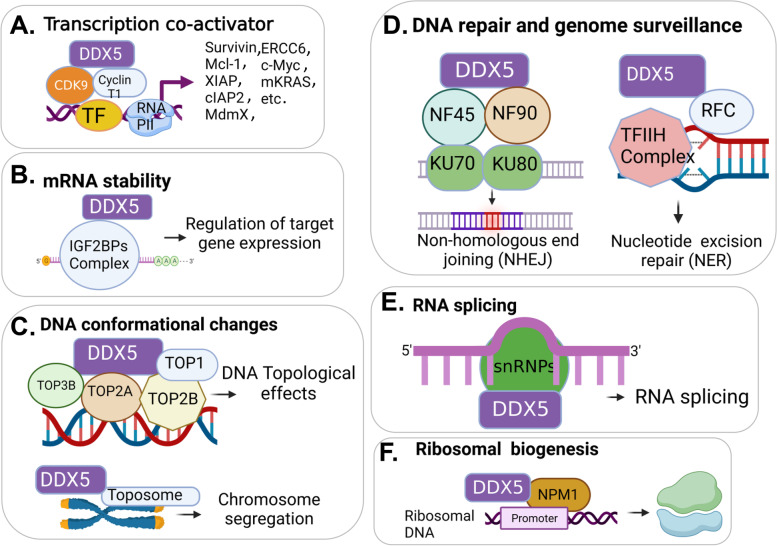
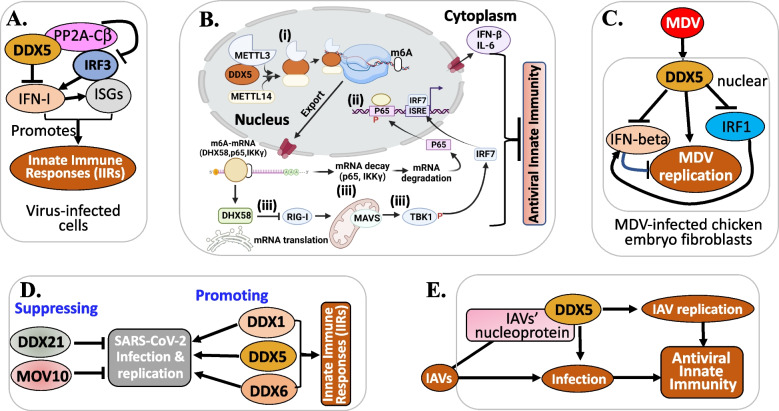
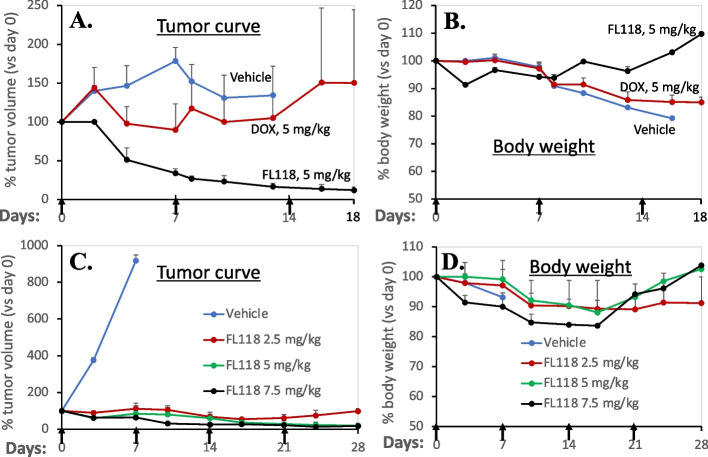
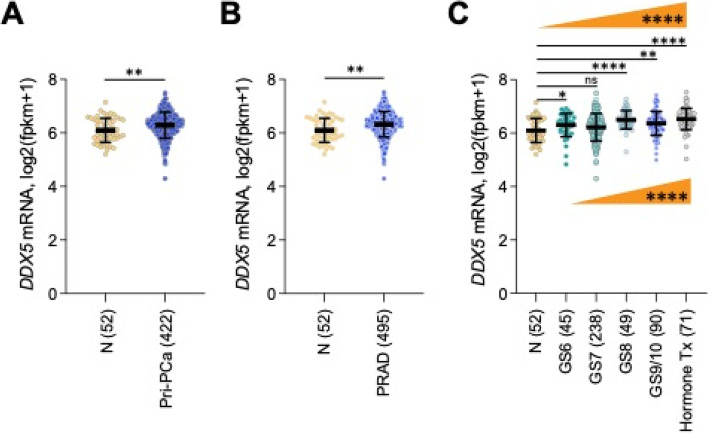
There is increasing evidence indicating the significant role of DDX5 (also called p68), acting as a master regulator and a potential biomarker and target, in tumorigenesis, proliferation, metastasis and treatment resistance for cancer therapy. However, DDX5 has also been reported to act as an oncosuppressor. These seemingly contradictory observations can be reconciled by DDX5's role in DNA repair. This is because cancer cell apoptosis and malignant transformation can represent the two possible outcomes of a single process regulated by DDX5, reflecting different intensity of DNA damage. Thus, targeting DDX5 could potentially shift cancer cells from a growth-arrested state (necessary for DNA repair) to apoptosis and cell killing. In addition to the increasingly recognized role of DDX5 in global genome stability surveillance and DNA damage repair, DDX5 has been implicated in multiple oncogenic signaling pathways. DDX5 appears to utilize distinct signaling cascades via interactions with unique proteins in different types of tissues/cells to elicit opposing roles (e.g., smooth muscle cells versus cancer cells). Such unique features make DDX5 an intriguing therapeutic target for the treatment of human cancers, with limited low toxicity to normal tissues. In this review, we discuss the multifaceted functions of DDX5 in DNA repair in cancer, immune suppression, oncogenic metabolic rewiring, virus infection promotion, and negative impact on the human microbiome (microbiota). We also provide new data showing that FL118, a molecular glue DDX5 degrader, selectively works against current treatment-resistant prostate cancer organoids/cells. Altogether, current studies demonstrate that DDX5 may represent a unique oncotarget for effectively conquering cancer with minimal toxicity to normal tissues.
Keywords: Cancer metabolic control; Cancer therapeutics; DDX5; DDX5 degrader FL118; DNA repair; Immune suppression; Microbiota negative influence; Oncotarget; Virus infection promotion; p68.
© 2023. Italian National Cancer Institute ‘Regina Elena’.
Conflict of interest statement
FL118 and FL118 core structure platform-based analogs will be further developed in Canget BioTekpharma LLC (
Figures







Similar articles
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
Interventions targeted at women to encourage the uptake of cervical screening.Cochrane Database Syst Rev. 2021 Sep 6;9(9):CD002834. doi: 10.1002/14651858.CD002834.pub3. Cochrane Database Syst Rev. 2021. PMID: 34694000 Free PMC article.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
[Volume and health outcomes: evidence from systematic reviews and from evaluation of Italian hospital data].Epidemiol Prev. 2013 Mar-Jun;37(2-3 Suppl 2):1-100. Epidemiol Prev. 2013. PMID: 23851286 Italian.
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
Cited by
-
Diverse roles of aldolase enzymes in cancer development, drug resistance and therapeutic approaches as moonlighting enzymes.Med Oncol. 2024 Aug 9;41(9):224. doi: 10.1007/s12032-024-02470-x. Med Oncol. 2024. PMID: 39120781 Review.
-
METTL1-modulated LSM14A facilitates proliferation and migration in glioblastoma via the stabilization of DDX5.iScience. 2024 Jun 8;27(7):110225. doi: 10.1016/j.isci.2024.110225. eCollection 2024 Jul 19. iScience. 2024. PMID: 39040050 Free PMC article.
-
Structure-Activity Relationship of FL118 Platform Position 7 Versus Position 9-Derived Compounds and Their Mechanism of Action and Antitumor Activity.J Med Chem. 2023 Dec 28;66(24):16888-16916. doi: 10.1021/acs.jmedchem.3c01589. Epub 2023 Dec 15. J Med Chem. 2023. PMID: 38100041 Free PMC article.
-
Biomaterial-assisted organoid technology for disease modeling and drug screening.Mater Today Bio. 2024 Dec 31;30:101438. doi: 10.1016/j.mtbio.2024.101438. eCollection 2025 Feb. Mater Today Bio. 2024. PMID: 39866785 Free PMC article. Review.
-
Sex Differences in a Novel Mouse Model of Spinocerebellar Ataxia Type 1 (SCA1).Int J Mol Sci. 2025 Mar 14;26(6):2623. doi: 10.3390/ijms26062623. Int J Mol Sci. 2025. PMID: 40141263 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
