

Conformational energies of reference organic molecules: benchmarking of common efficient computational methods against coupled cluster theory
- PMID: 37597063
- PMCID: PMC10618395
- DOI: 10.1007/s10822-023-00513-5
Conformational energies of reference organic molecules: benchmarking of common efficient computational methods against coupled cluster theory
Erratum in
-
Correction to: Conformational energies of reference organic molecules: benchmarking of common efficient computational methods against coupled cluster theory.J Comput Aided Mol Des. 2023 Dec;37(12):657. doi: 10.1007/s10822-023-00531-3. J Comput Aided Mol Des. 2023. PMID: 37773475 Free PMC article. No abstract available.
Abstract
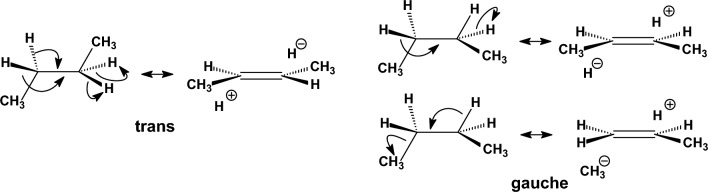
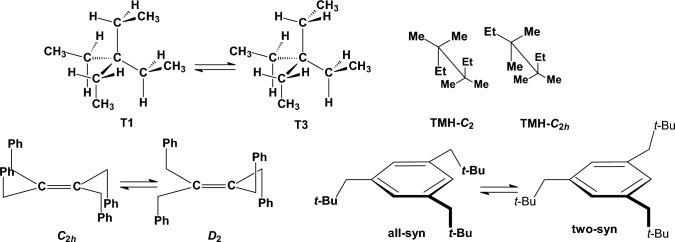
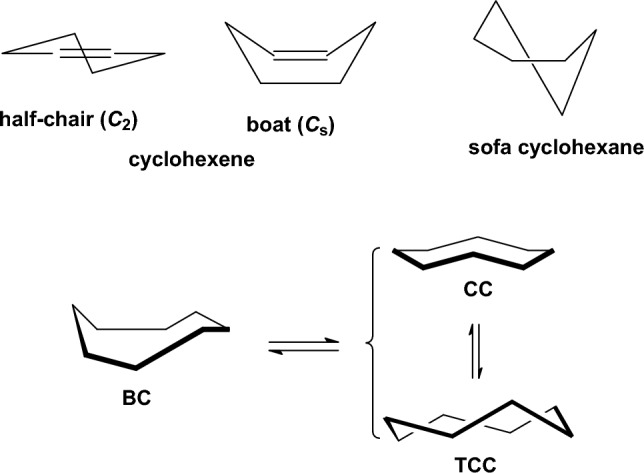
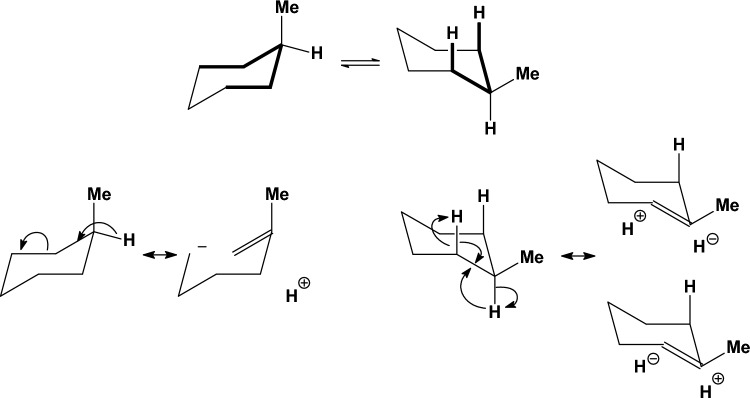
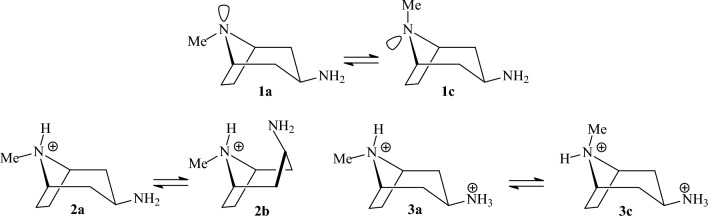
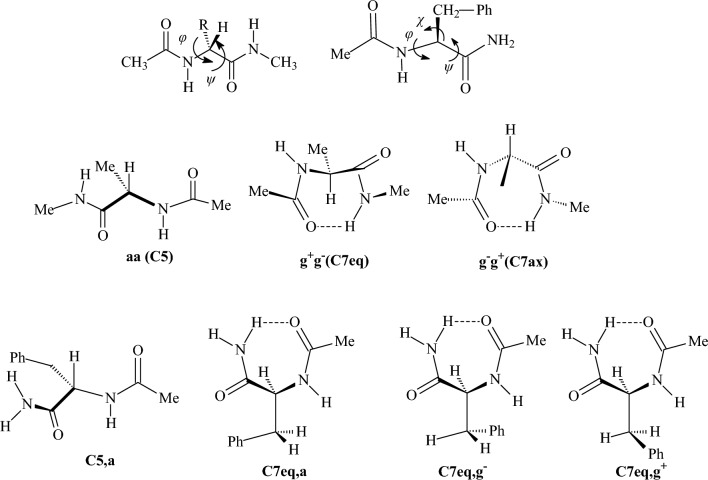
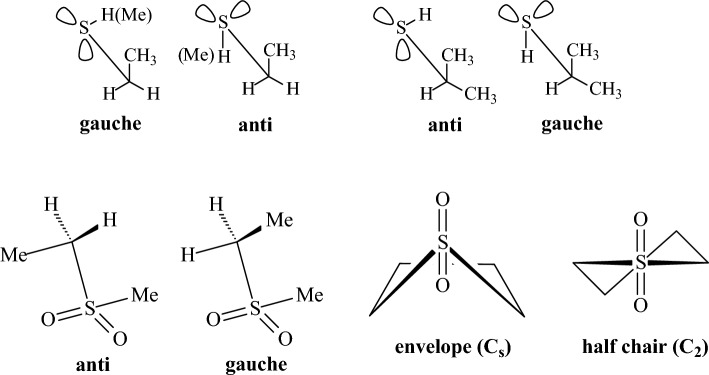
We selected 145 reference organic molecules that include model fragments used in computer-aided drug design. We calculated 158 conformational energies and barriers using force fields, with wide applicability in commercial and free softwares and extensive application on the calculation of conformational energies of organic molecules, e.g. the UFF and DREIDING force fields, the Allinger's force fields MM3-96, MM3-00, MM4-8, the MM2-91 clones MMX and MM+, the MMFF94 force field, MM4, ab initio Hartree-Fock (HF) theory with different basis sets, the standard density functional theory B3LYP, the second-order post-HF MP2 theory and the Domain-based Local Pair Natural Orbital Coupled Cluster DLPNO-CCSD(T) theory, with the latter used for accurate reference values. The data set of the organic molecules includes hydrocarbons, haloalkanes, conjugated compounds, and oxygen-, nitrogen-, phosphorus- and sulphur-containing compounds. We reviewed in detail the conformational aspects of these model organic molecules providing the current understanding of the steric and electronic factors that determine the stability of low energy conformers and the literature including previous experimental observations and calculated findings. While progress on the computer hardware allows the calculations of thousands of conformations for later use in drug design projects, this study is an update from previous classical studies that used, as reference values, experimental ones using a variety of methods and different environments. The lowest mean error against the DLPNO-CCSD(T) reference was calculated for MP2 (0.35 kcal mol-1), followed by B3LYP (0.69 kcal mol-1) and the HF theories (0.81-1.0 kcal mol-1). As regards the force fields, the lowest errors were observed for the Allinger's force fields MM3-00 (1.28 kcal mol-1), ΜΜ3-96 (1.40 kcal mol-1) and the Halgren's MMFF94 force field (1.30 kcal mol-1) and then for the MM2-91 clones MMX (1.77 kcal mol-1) and MM+ (2.01 kcal mol-1) and MM4 (2.05 kcal mol-1). The DREIDING (3.63 kcal mol-1) and UFF (3.77 kcal mol-1) force fields have the lowest performance. These model organic molecules we used are often present as fragments in drug-like molecules. The values calculated using DLPNO-CCSD(T) make up a valuable data set for further comparisons and for improved force field parameterization.
Keywords: B3LYP; Conformational energies; DLPNO-CCSD(T); Force fields; Organic molecules.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing financial interest.
Figures













References
-
- Mohamadi F, Richards NGJ, Guida WC, et al. Macromodel? An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11(4):440–467. doi: 10.1002/jcc.540110405. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
