

Certain heterozygous variants in the kinase domain of the serine/threonine kinase NEK8 can cause an autosomal dominant form of polycystic kidney disease
- PMID: 37598857
- PMCID: PMC10592035
- DOI: 10.1016/j.kint.2023.07.021
Certain heterozygous variants in the kinase domain of the serine/threonine kinase NEK8 can cause an autosomal dominant form of polycystic kidney disease
Abstract
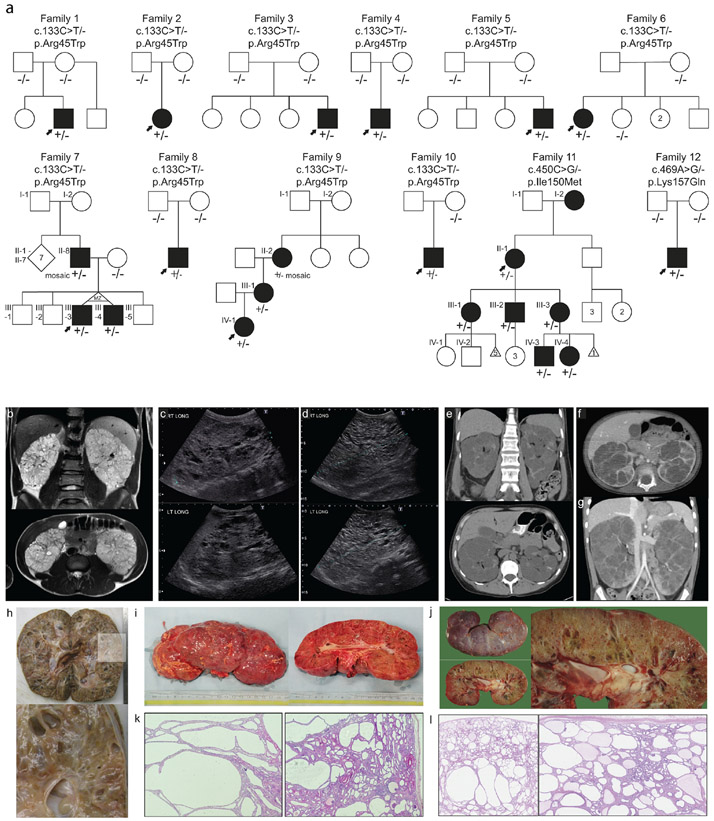
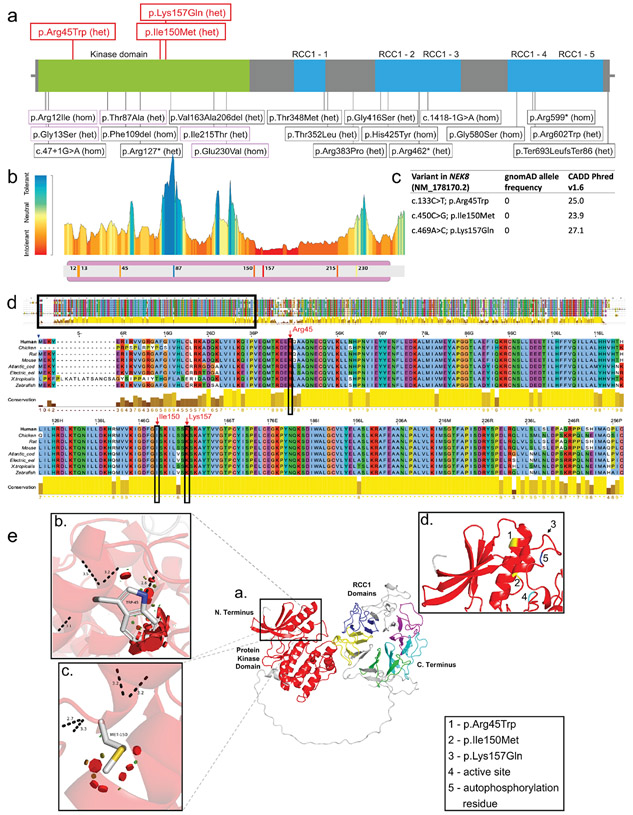
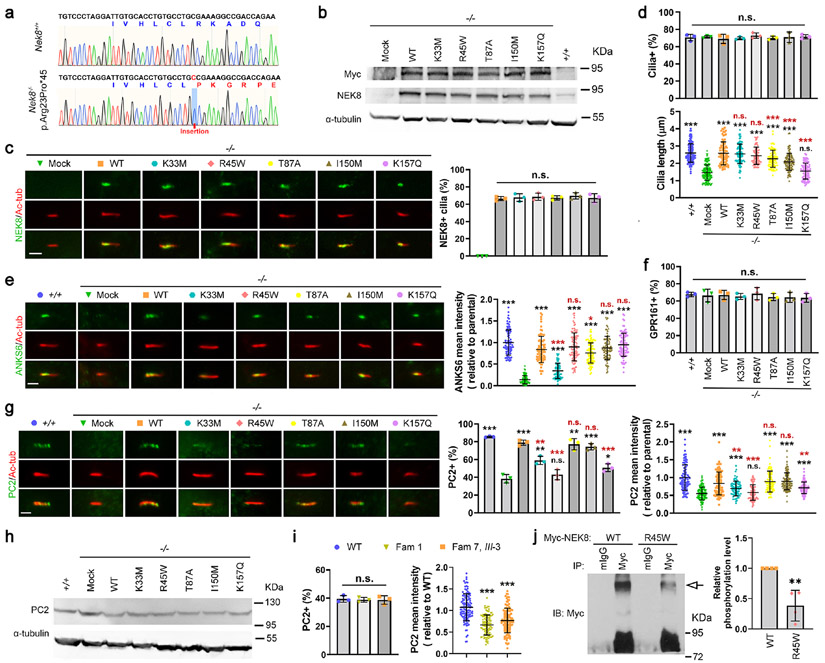
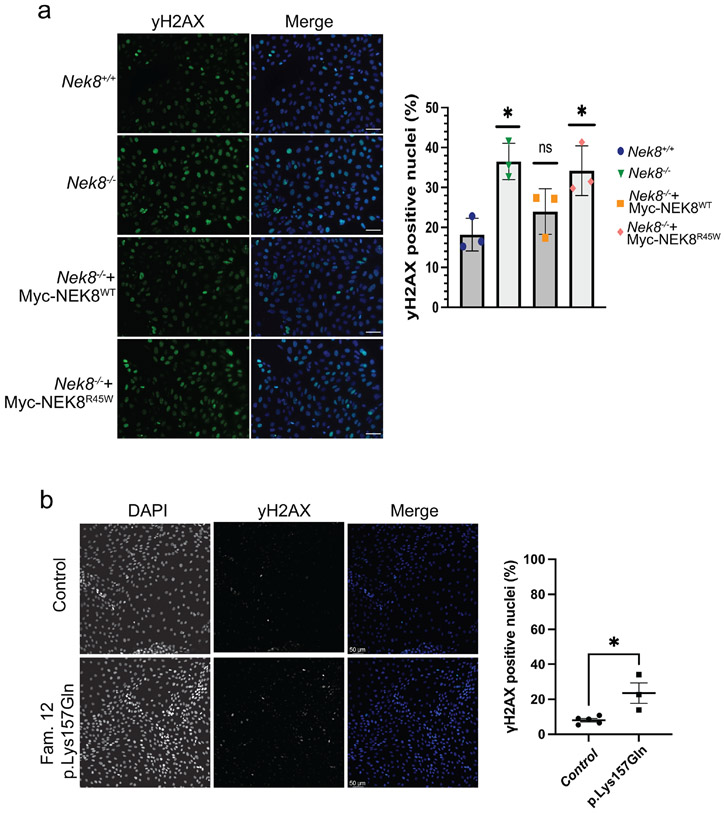
Autosomal dominant polycystic kidney disease (ADPKD) resulting from pathogenic variants in PKD1 and PKD2 is the most common form of PKD, but other genetic causes tied to primary cilia function have been identified. Biallelic pathogenic variants in the serine/threonine kinase NEK8 cause a syndromic ciliopathy with extra-kidney manifestations. Here we identify NEK8 as a disease gene for ADPKD in 12 families. Clinical evaluation was combined with functional studies using fibroblasts and tubuloids from affected individuals. Nek8 knockout mouse kidney epithelial (IMCD3) cells transfected with wild type or variant NEK8 were further used to study ciliogenesis, ciliary trafficking, kinase function, and DNA damage responses. Twenty-one affected monoallelic individuals uniformly exhibited cystic kidney disease (mostly neonatal) without consistent extra-kidney manifestations. Recurrent de novo mutations of the NEK8 missense variant p.Arg45Trp, including mosaicism, were seen in ten families. Missense variants elsewhere within the kinase domain (p.Ile150Met and p.Lys157Gln) were also identified. Functional studies demonstrated normal localization of the NEK8 protein to the proximal cilium and no consistent cilia formation defects in patient-derived cells. NEK8-wild type protein and all variant forms of the protein expressed in Nek8 knockout IMCD3 cells were localized to cilia and supported ciliogenesis. However, Nek8 knockout IMCD3 cells expressing NEK8-p.Arg45Trp and NEK8-p.Lys157Gln showed significantly decreased polycystin-2 but normal ANKS6 localization in cilia. Moreover, p.Arg45Trp NEK8 exhibited reduced kinase activity in vitro. In patient derived tubuloids and IMCD3 cells expressing NEK8-p.Arg45Trp, DNA damage signaling was increased compared to healthy passage-matched controls. Thus, we propose a dominant-negative effect for specific heterozygous missense variants in the NEK8 kinase domain as a new cause of PKD.
Keywords: NEK8; ciliopathy; kinase; polycystic kidney disease.
Copyright © 2023 International Society of Nephrology. All rights reserved.
Conflict of interest statement
Declaration of interests
The authors declare no competing interests. Jennifer Stallworth completed this work as an employee at GGC, and is now a Sanofi employee.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
