

Development of a knowledge graph framework to ease and empower translational approaches in plant research: a use-case on grain legumes
- PMID: 37601035
- PMCID: PMC10435283
- DOI: 10.3389/frai.2023.1191122
Development of a knowledge graph framework to ease and empower translational approaches in plant research: a use-case on grain legumes
Abstract
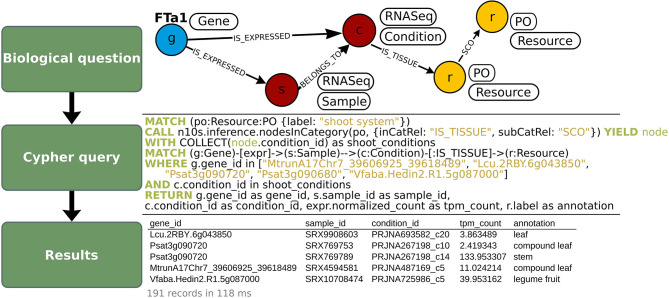
While the continuing decline in genotyping and sequencing costs has largely benefited plant research, some key species for meeting the challenges of agriculture remain mostly understudied. As a result, heterogeneous datasets for different traits are available for a significant number of these species. As gene structures and functions are to some extent conserved through evolution, comparative genomics can be used to transfer available knowledge from one species to another. However, such a translational research approach is complex due to the multiplicity of data sources and the non-harmonized description of the data. Here, we provide two pipelines, referred to as structural and functional pipelines, to create a framework for a NoSQL graph-database (Neo4j) to integrate and query heterogeneous data from multiple species. We call this framework Orthology-driven knowledge base framework for translational research (Ortho_KB). The structural pipeline builds bridges across species based on orthology. The functional pipeline integrates biological information, including QTL, and RNA-sequencing datasets, and uses the backbone from the structural pipeline to connect orthologs in the database. Queries can be written using the Neo4j Cypher language and can, for instance, lead to identify genes controlling a common trait across species. To explore the possibilities offered by such a framework, we populated Ortho_KB to obtain OrthoLegKB, an instance dedicated to legumes. The proposed model was evaluated by studying the conservation of a flowering-promoting gene. Through a series of queries, we have demonstrated that our knowledge graph base provides an intuitive and powerful platform to support research and development programmes.
Keywords: OrthoLegKB; Ortho_KB; comparative omics; gene expression; graph database; ontology; orthology; quantitative genetics.
Copyright © 2023 Imbert, Kreplak, Flores, Aubert, Burstin and Tayeh.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures







References
-
- Abuoda G., Dell'Aglio D., Keen A., Hose K. (2022). Transforming RDF-star to property graphs: A preliminary analysis of transformation approaches – extended version. arXiv [Preprint]. arXiv: 2210.05781. 10.48550/arXiv.2210.05781 - DOI
-
- Bandi V., Gutwin C. (2020). Interactive Exploration of Genomic Conservation in Proceedings of Graphics Interface 2020 GI 2020. Toronto: Canadian Human-Computer Communications Society/Société canadienne du dialogue humain-machine, 74–83.
-
- Barrasa J. (2022). Neosemantics (n10s). Available online at: https://github.com/neo4j-labs/neosemantics (accessed December 21, 2022).
-
- Berardini T. Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., et al. (2015). The arabidopsis information resource: making and mining the “gold standard” annotated reference plant genome: tair: making and mining the “gold standard” plant genome. Genesis 53, 474–485. 10.1002/dvg.22877 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous