

Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes
- PMID: 37601314
- PMCID: PMC10439824
- DOI: 10.1002/aps3.11533
Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes
Erratum in
-
Correction to Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes.Appl Plant Sci. 2023 Nov 7;11(6):e11553. doi: 10.1002/aps3.11553. eCollection 2023 Nov-Dec. Appl Plant Sci. 2023. PMID: 38106536 Free PMC article.
Abstract
Premise: Robust standards to evaluate quality and completeness are lacking in eukaryotic structural genome annotation, as genome annotation software is developed using model organisms and typically lacks benchmarking to comprehensively evaluate the quality and accuracy of the final predictions. The annotation of plant genomes is particularly challenging due to their large sizes, abundant transposable elements, and variable ploidies. This study investigates the impact of genome quality, complexity, sequence read input, and method on protein-coding gene predictions.
Methods: The impact of repeat masking, long-read and short-read inputs, and de novo and genome-guided protein evidence was examined in the context of the popular BRAKER and MAKER workflows for five plant genomes. The annotations were benchmarked for structural traits and sequence similarity.
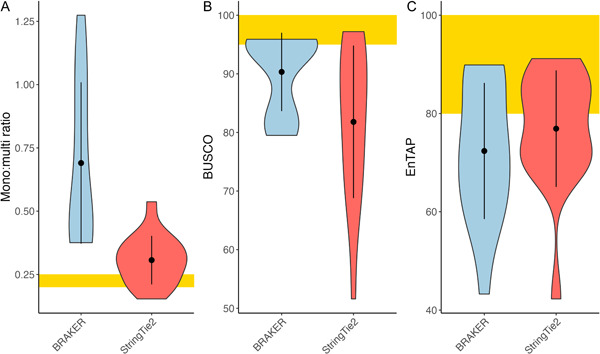
Results: Benchmarks that reflect gene structures, reciprocal similarity search alignments, and mono-exonic/multi-exonic gene counts provide a more complete view of annotation accuracy. Transcripts derived from RNA-read alignments alone are not sufficient for genome annotation. Gene prediction workflows that combine evidence-based and ab initio approaches are recommended, and a combination of short and long reads can improve genome annotation. Adding protein evidence from de novo assemblies, genome-guided transcriptome assemblies, or full-length proteins from OrthoDB generates more putative false positives as implemented in the current workflows. Post-processing with functional and structural filters is highly recommended.
Discussion: While the annotation of non-model plant genomes remains complex, this study provides recommendations for inputs and methodological approaches. We discuss a set of best practices to generate an optimal plant genome annotation and present a more robust set of metrics to evaluate the resulting predictions.
Keywords: BRAKER; MAKER; StringTie2; TSEBRA; gene identification; genome annotation; plant genomes.
© 2023 The Authors. Applications in Plant Sciences published by Wiley Periodicals LLC on behalf of Botanical Society of America.
Figures




Similar articles
-
A long-read and short-read transcriptomics approach provides the first high-quality reference transcriptome and genome annotation for Pseudotsuga menziesii (Douglas-fir).G3 (Bethesda). 2023 Feb 9;13(2):jkac304. doi: 10.1093/g3journal/jkac304. G3 (Bethesda). 2023. PMID: 36454025 Free PMC article.
-
TSEBRA: transcript selector for BRAKER.BMC Bioinformatics. 2021 Nov 25;22(1):566. doi: 10.1186/s12859-021-04482-0. BMC Bioinformatics. 2021. PMID: 34823473 Free PMC article.
-
Illuminating the dark side of the human transcriptome with long read transcript sequencing.BMC Genomics. 2020 Oct 30;21(1):751. doi: 10.1186/s12864-020-07123-7. BMC Genomics. 2020. PMID: 33126848 Free PMC article.
-
A simple guide to de novo transcriptome assembly and annotation.Brief Bioinform. 2022 Mar 10;23(2):bbab563. doi: 10.1093/bib/bbab563. Brief Bioinform. 2022. PMID: 35076693 Free PMC article. Review.
-
Genome annotation: From human genetics to biodiversity genomics.Cell Genom. 2023 Aug 1;3(8):100375. doi: 10.1016/j.xgen.2023.100375. eCollection 2023 Aug 9. Cell Genom. 2023. PMID: 37601977 Free PMC article. Review.
Cited by
-
Galba: genome annotation with miniprot and AUGUSTUS.BMC Bioinformatics. 2023 Aug 31;24(1):327. doi: 10.1186/s12859-023-05449-z. BMC Bioinformatics. 2023. PMID: 37653395 Free PMC article.
-
Assembly and annotation of the black spruce genome provide insights on spruce phylogeny and evolution of stress response.G3 (Bethesda). 2023 Dec 29;14(1):jkad247. doi: 10.1093/g3journal/jkad247. G3 (Bethesda). 2023. PMID: 37875130 Free PMC article.
-
Crossroads of assembling a moss genome: navigating contaminants and horizontal gene transfer in the moss Physcomitrellopsis africana.G3 (Bethesda). 2024 Jul 8;14(7):jkae104. doi: 10.1093/g3journal/jkae104. G3 (Bethesda). 2024. PMID: 38781445 Free PMC article.
-
Annotation of protein-coding genes in 49 diatom genomes from the Bacillariophyta clade.Sci Data. 2025 Jun 11;12(1):985. doi: 10.1038/s41597-025-05306-z. Sci Data. 2025. PMID: 40500266 Free PMC article.
-
GALBA: Genome Annotation with Miniprot and AUGUSTUS.bioRxiv [Preprint]. 2023 Apr 10:2023.04.10.536199. doi: 10.1101/2023.04.10.536199. bioRxiv. 2023. Update in: BMC Bioinformatics. 2023 Aug 31;24(1):327. doi: 10.1186/s12859-023-05449-z. PMID: 37090650 Free PMC article. Updated. Preprint.
References
-
- Andrews, S. 2010. FastQC: A quality control tool for high throughput sequence data. Available online. Website: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [accessed 17 May 2018].
-
- Arabidopsis Genome Initiative . 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature 408(6814): 796–815. - PubMed
