

Assessing the evolution of SARS-CoV-2 lineages and the dynamic associations between nucleotide variations
- PMID: 37601437
- PMCID: PMC10436015
- DOI: 10.1099/acmi.0.000513.v3
Assessing the evolution of SARS-CoV-2 lineages and the dynamic associations between nucleotide variations
Abstract
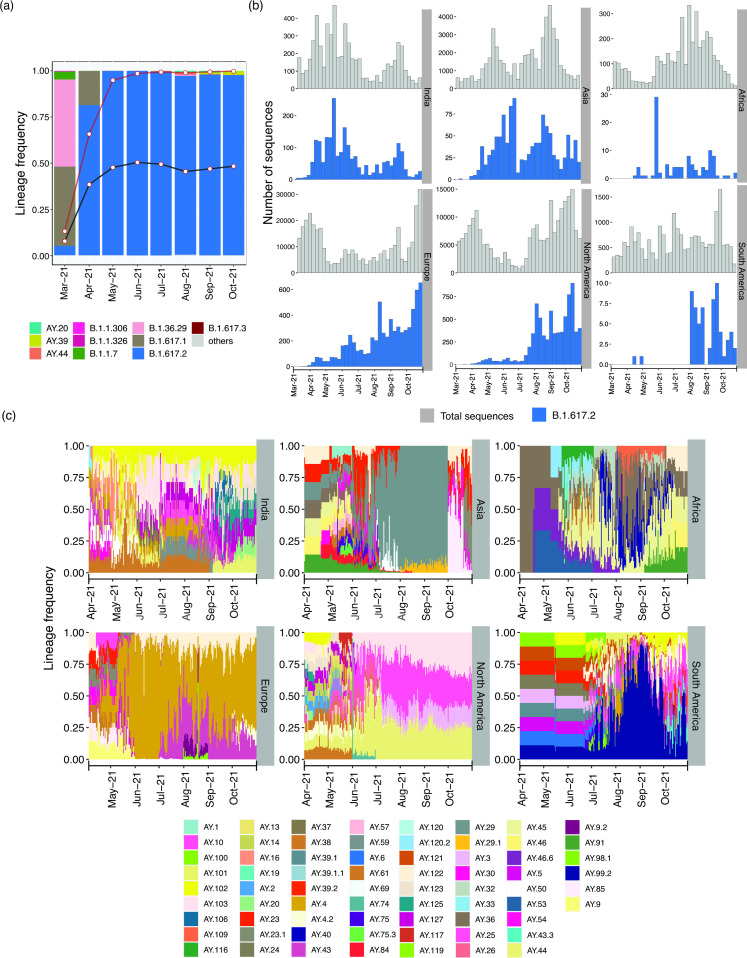
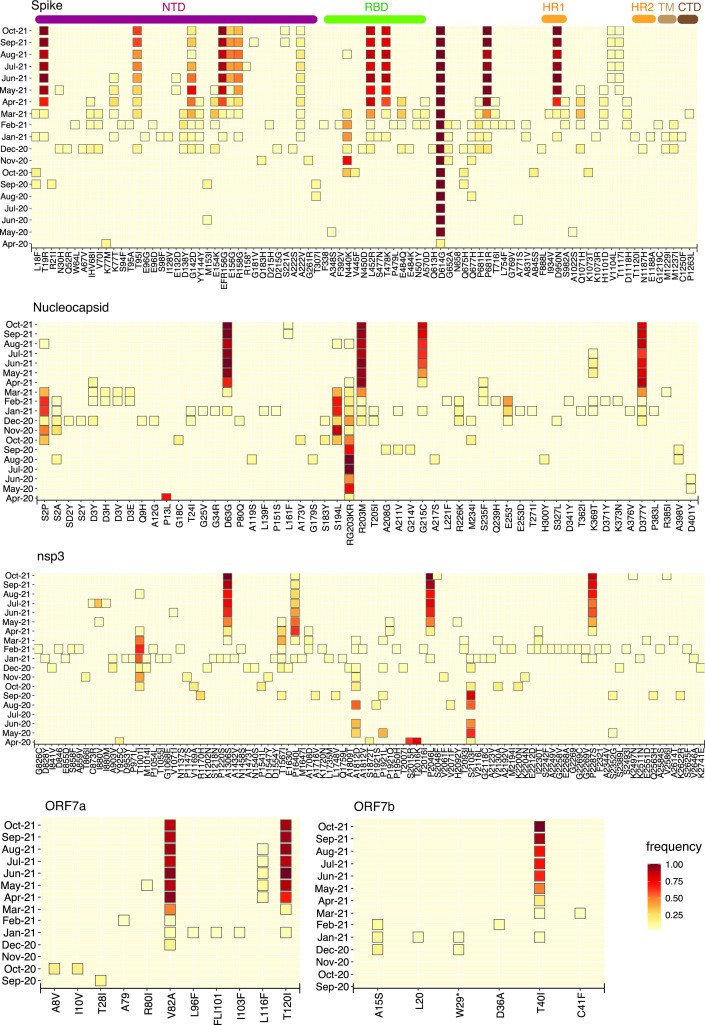
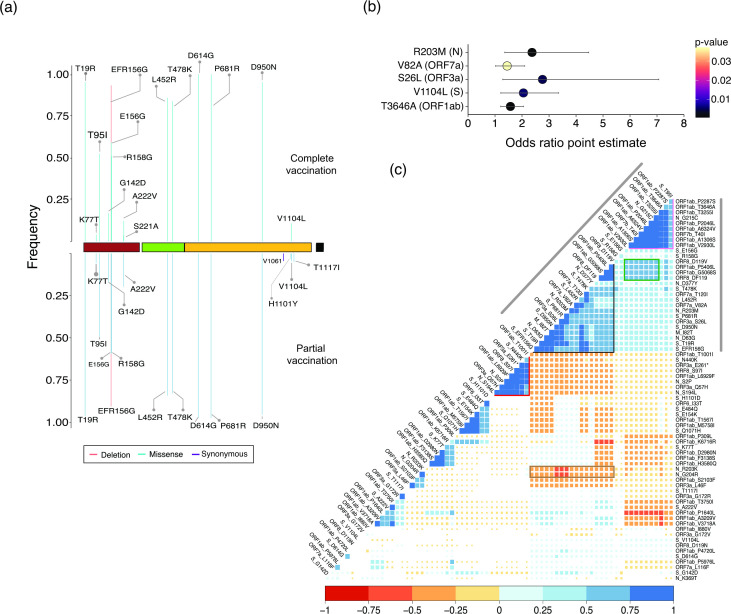
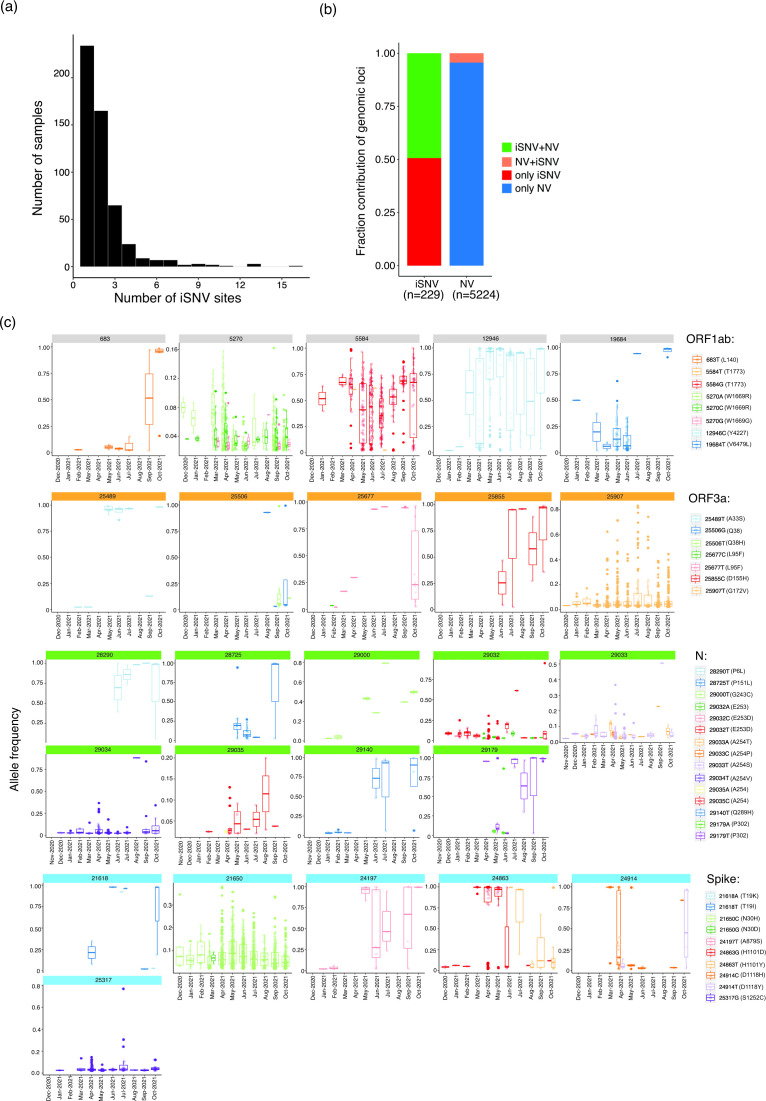
Despite seminal advances towards understanding the infection mechanism of SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), it continues to cause significant morbidity and mortality worldwide. Though mass immunization programmes have been implemented in several countries, the viral transmission cycle has shown a continuous progression in the form of multiple waves. A constant change in the frequencies of dominant viral lineages, arising from the accumulation of nucleotide variations (NVs) through favourable selection, is understandably expected to be a major determinant of disease severity and possible vaccine escape. Indeed, worldwide efforts have been initiated to identify specific virus lineage(s) and/or NVs that may cause a severe clinical presentation or facilitate vaccination breakthrough. Since host genetics is expected to play a major role in shaping virus evolution, it is imperative to study the role of genome-wide SARS-CoV-2 NVs across various populations. In the current study, we analysed the whole genome sequence of 3543 SARS-CoV-2-infected samples obtained from the state of Telangana, India (including 210 from our previous study), collected over an extended period from April 2020 to October 2021. We present a unique perspective on the evolution of prevalent virus lineages and NVs during this period. We also highlight the presence of specific NVs likely to be associated favourably with samples classified as vaccination breakthroughs. Finally, we report genome-wide intra-host variations at novel genomic positions. The results presented here provide critical insights into virus evolution over an extended period and pave the way to rigorously investigate the role of specific NVs in vaccination breakthroughs.
Keywords: COVID-19; SARS-CoV-2 genome evolution; mutational co-occurrence; pathogenesis; vaccination breakthrough.
© 2023 The Authors.
Conflict of interest statement
The author(s) declare that there are no conflicts of interest.
Figures
References
-
- Frampton D, Rampling T, Cross A, Bailey H, Heaney J, et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: a whole-genome sequencing and hospital-based cohort study. Lancet Infect Dis. 2021;21:1246–1256. doi: 10.1016/S1473-3099(21)00170-5. - DOI - PMC - PubMed
Associated data
LinkOut - more resources
Full Text Sources
Miscellaneous