

Targeting MCL-1 protein to treat cancer: opportunities and challenges
- PMID: 37601693
- PMCID: PMC10436212
- DOI: 10.3389/fonc.2023.1226289
Targeting MCL-1 protein to treat cancer: opportunities and challenges
Abstract
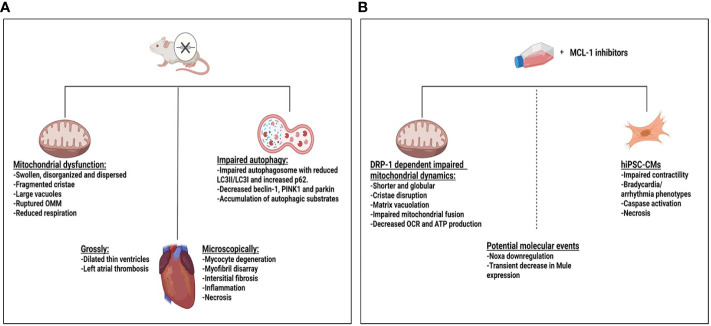
Evading apoptosis has been linked to tumor development and chemoresistance. One mechanism for this evasion is the overexpression of prosurvival B-cell lymphoma-2 (BCL-2) family proteins, which gives cancer cells a survival advantage. Mcl-1, a member of the BCL-2 family, is among the most frequently amplified genes in cancer. Targeting myeloid cell leukemia-1 (MCL-1) protein is a successful strategy to induce apoptosis and overcome tumor resistance to chemotherapy and targeted therapy. Various strategies to inhibit the antiapoptotic activity of MCL-1 protein, including transcription, translation, and the degradation of MCL-1 protein, have been tested. Neutralizing MCL-1's function by targeting its interactions with other proteins via BCL-2 interacting mediator (BIM)S2A has been shown to be an equally effective approach. Encouraged by the design of venetoclax and its efficacy in chronic lymphocytic leukemia, scientists have developed other BCL-2 homology (BH3) mimetics-particularly MCL-1 inhibitors (MCL-1i)-that are currently in clinical trials for various cancers. While extensive reviews of MCL-1i are available, critical analyses focusing on the challenges of MCL-1i and their optimization are lacking. In this review, we discuss the current knowledge regarding clinically relevant MCL-1i and focus on predictive biomarkers of response, mechanisms of resistance, major issues associated with use of MCL-1i, and the future use of and maximization of the benefits from these agents.
Keywords: BCL-2 family; BCL-2 family proteins; MCL-1 inhibitors; MCL-1 protein; apoptosis; cancer therapy.
Copyright © 2023 Tantawy, Timofeeva, Sarkar and Gandhi.
Conflict of interest statement
Author VG has received research grants from Pharmacyclics, Acerta, Gilead, Sunesis, ClearCreek Bio, Infinity, and Loxo Oncology. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources