

Understanding the host-pathogen evolutionary balance through Gaussian process modeling of SARS-CoV-2
- PMID: 37602209
- PMCID: PMC10436005
- DOI: 10.1016/j.patter.2023.100800
Understanding the host-pathogen evolutionary balance through Gaussian process modeling of SARS-CoV-2
Abstract
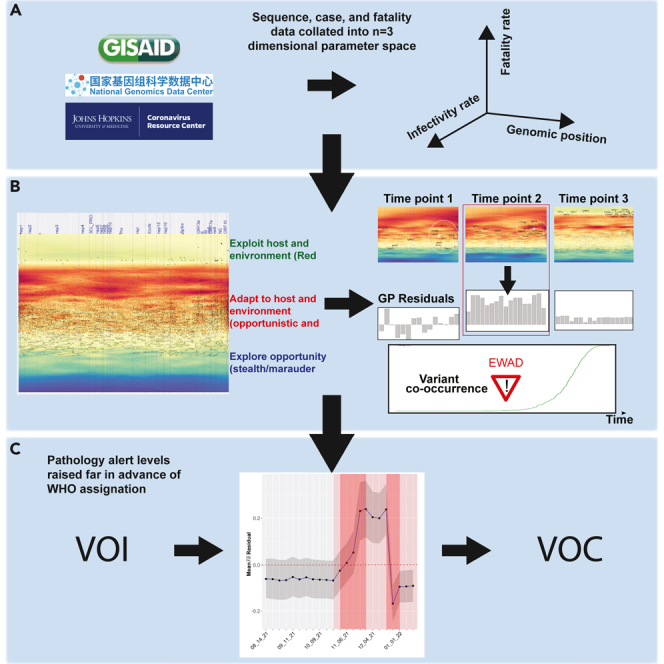
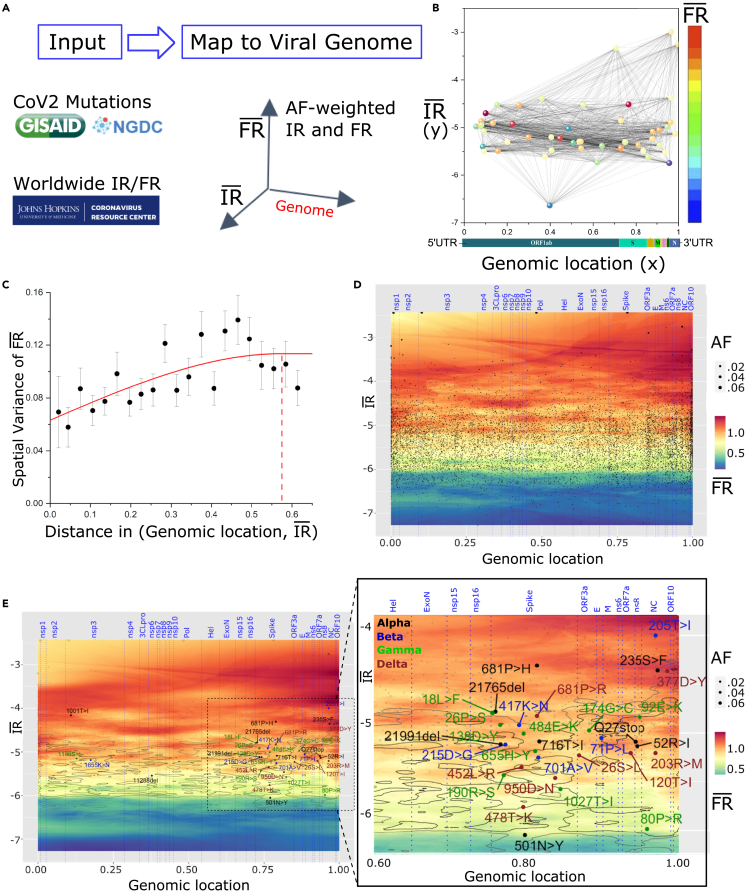
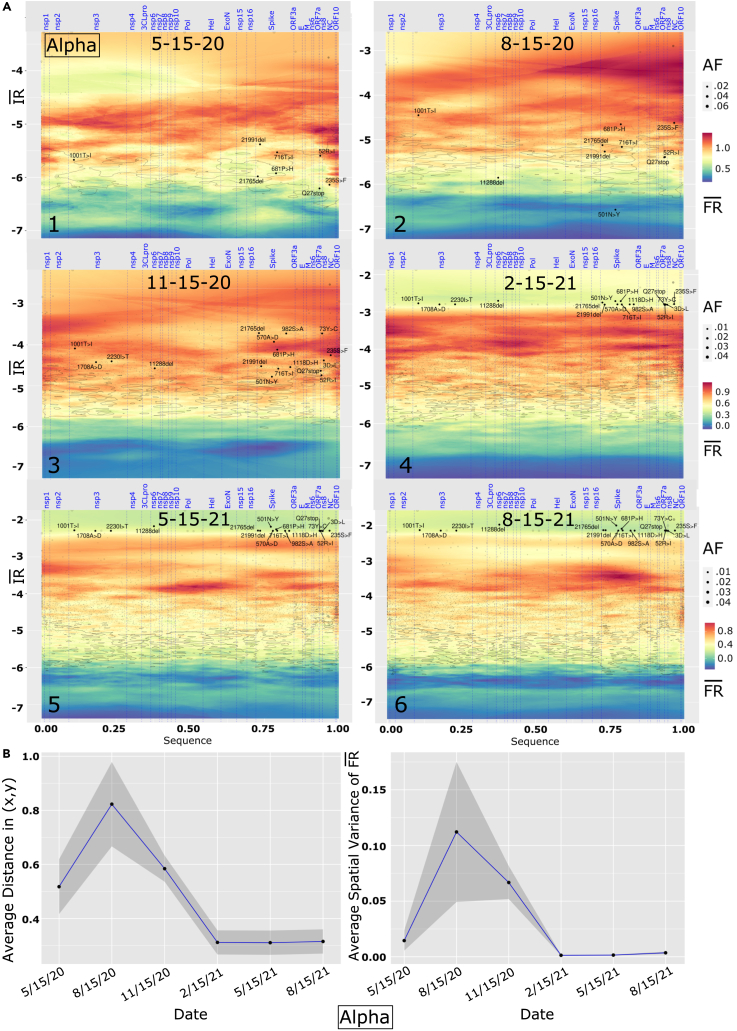
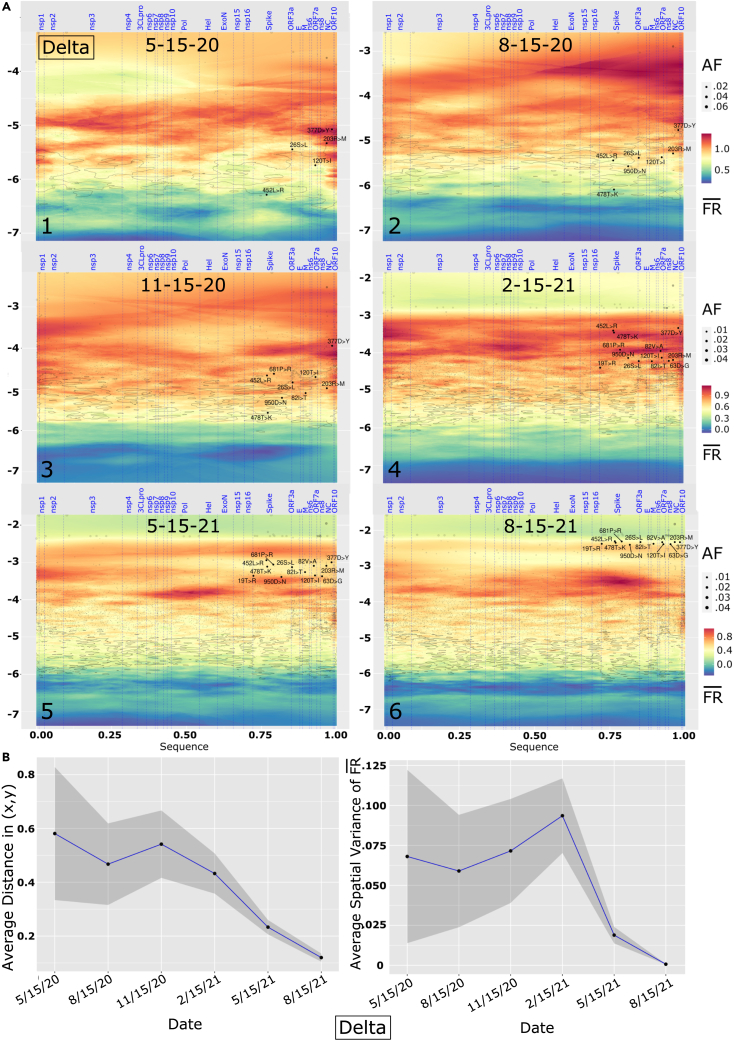
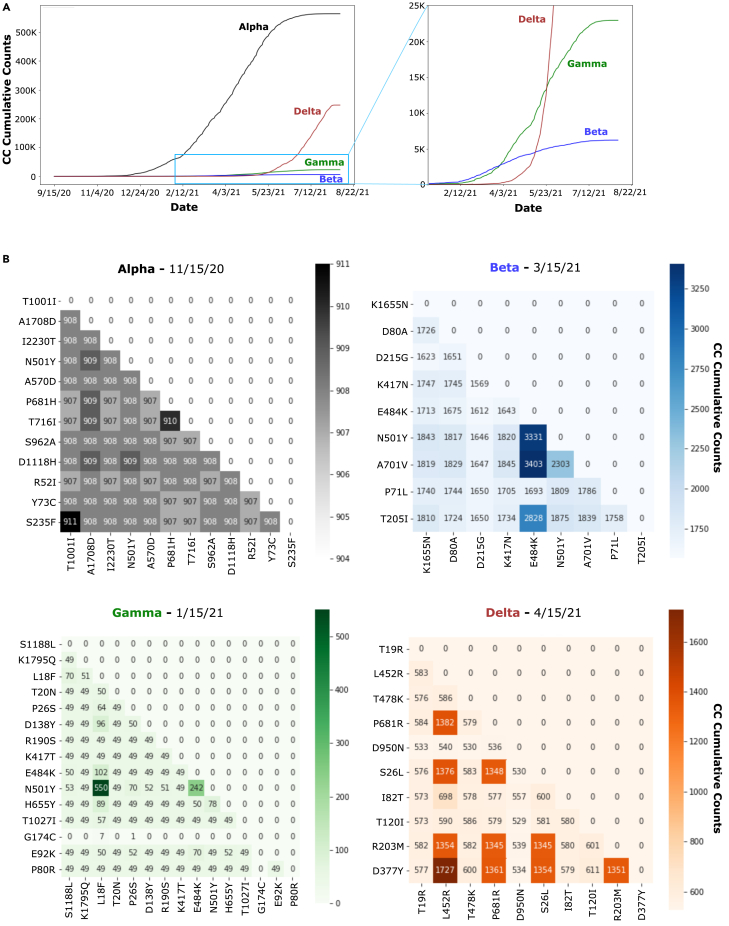
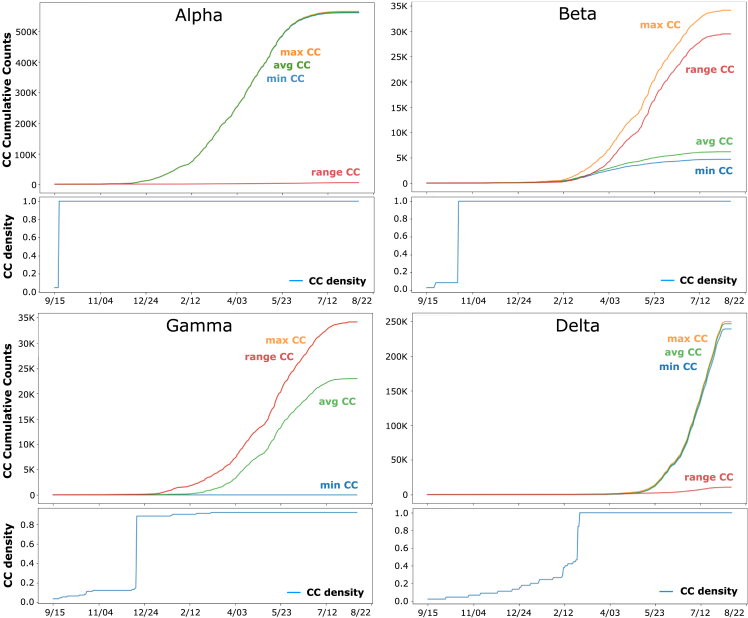
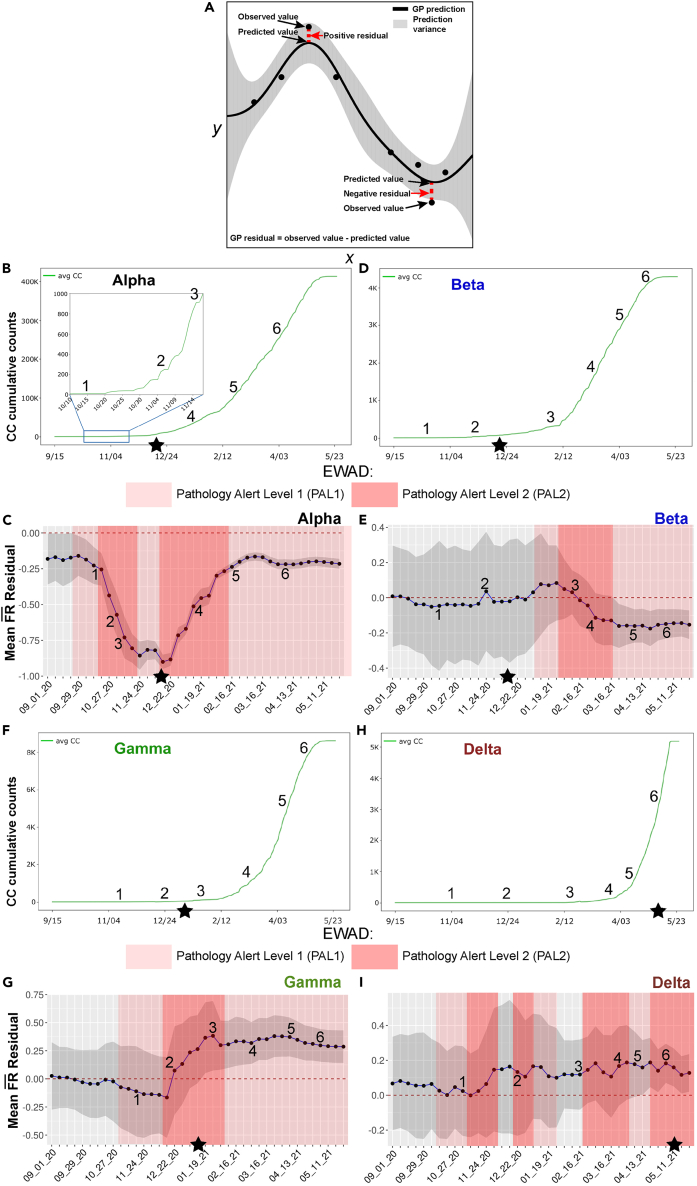
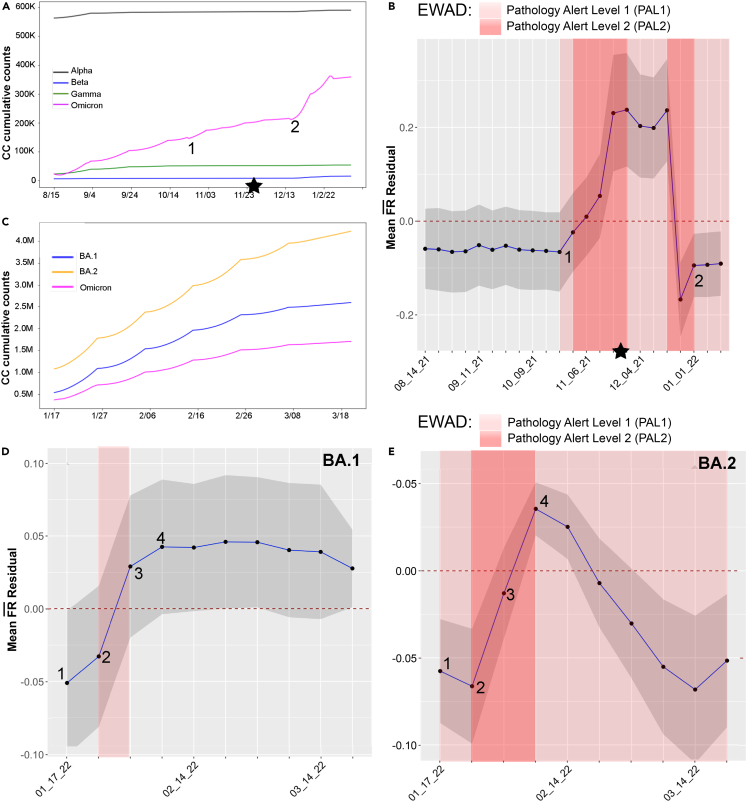
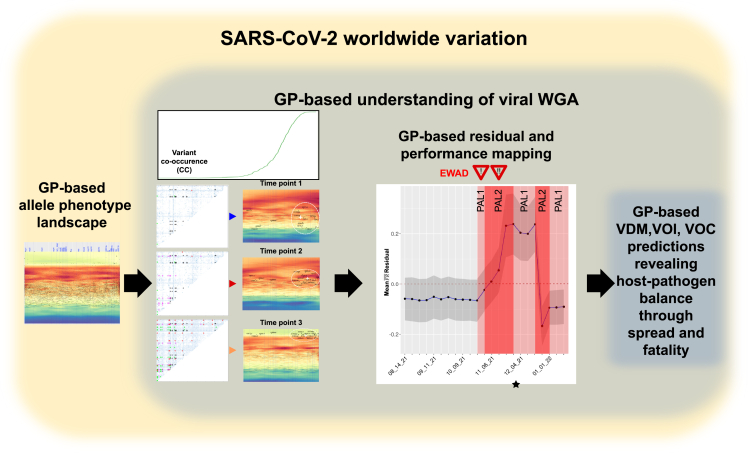
We have developed a machine learning (ML) approach using Gaussian process (GP)-based spatial covariance (SCV) to track the impact of spatial-temporal mutational events driving host-pathogen balance in biology. We show how SCV can be applied to understanding the response of evolving covariant relationships linking the variant pattern of virus spread to pathology for the entire SARS-CoV-2 genome on a daily basis. We show that GP-based SCV relationships in conjunction with genome-wide co-occurrence analysis provides an early warning anomaly detection (EWAD) system for the emergence of variants of concern (VOCs). EWAD can anticipate changes in the pattern of performance of spread and pathology weeks in advance, identifying signatures destined to become VOCs. GP-based analyses of variation across entire viral genomes can be used to monitor micro and macro features responsible for host-pathogen balance. The versatility of GP-based SCV defines starting point for understanding nature's evolutionary path to complexity through natural selection.
Keywords: Gaussian processes; SARS-CoV-2; early warning; evolution; genomic surveillance; host-pathogen; machine learning; spatial covariance.
© 2023.
Conflict of interest statement
The authors declare no competing interests. The authors declare no advisory, management or consulting positions. C.W. and W.E.B. have filed a patent application for the SCV methodology (serial no. US2021/0324474). C.W. and W.E.B. have filed a PCT application (serial no. PCT/US2022/039594) for VarC methodology.
Figures
References
-
- WHO Coronavirus (COVID-19) Dashboard. (2022). https://covid19.who.int.
-
- Levin A.T., Hanage W.P., Owusu-Boaitey N., Cochran K.B., Walsh S.P., Meyerowitz-Katz G. Assessing the age specificity of infection fatality rates for COVID-19: systematic review, meta-analysis, and public policy implications. Eur. J. Epidemiol. 2020;35:1123–1138. doi: 10.1007/s10654-020-00698-1. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
