

Genetic analysis of blood molecular phenotypes reveals common properties in the regulatory networks affecting complex traits
- PMID: 37604891
- PMCID: PMC10442420
- DOI: 10.1038/s41467-023-40569-3
Genetic analysis of blood molecular phenotypes reveals common properties in the regulatory networks affecting complex traits
Abstract
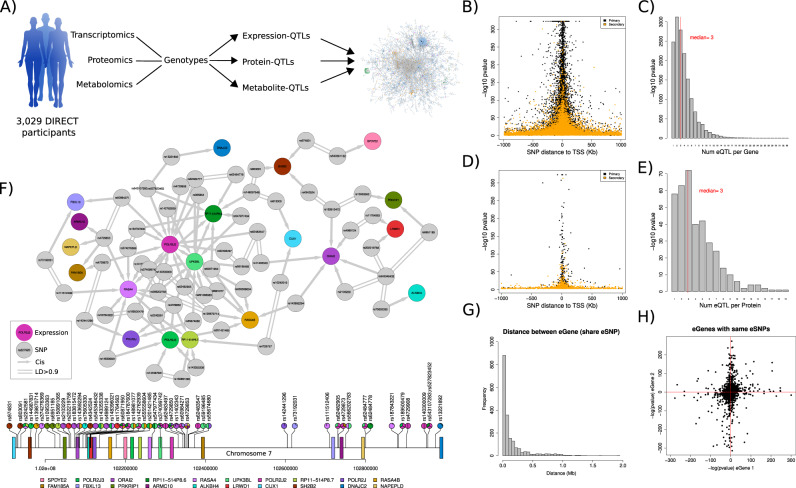
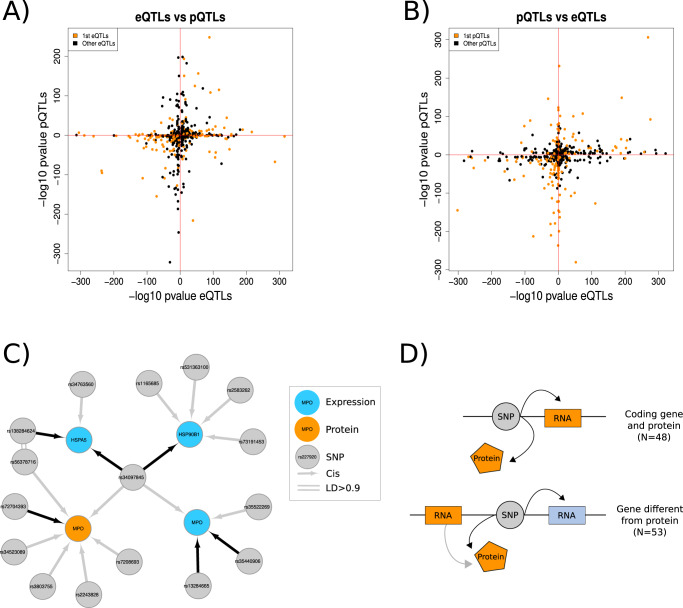
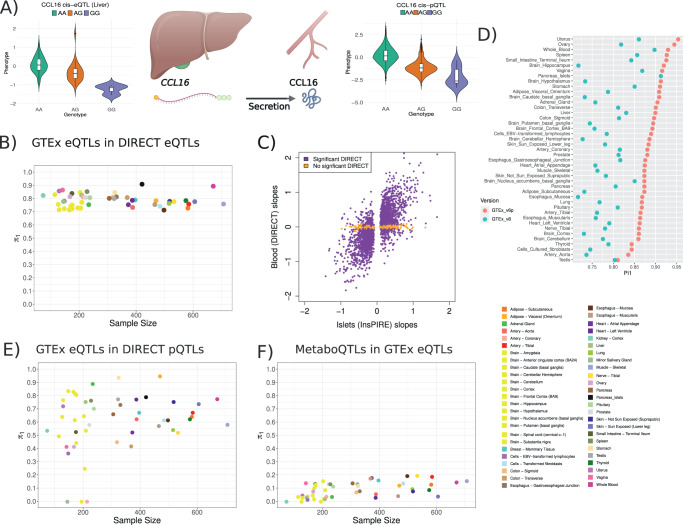
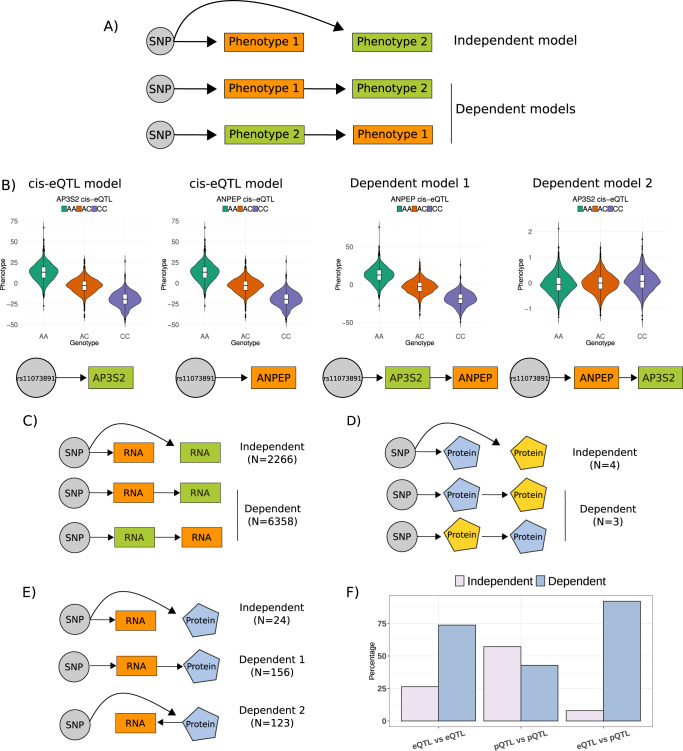
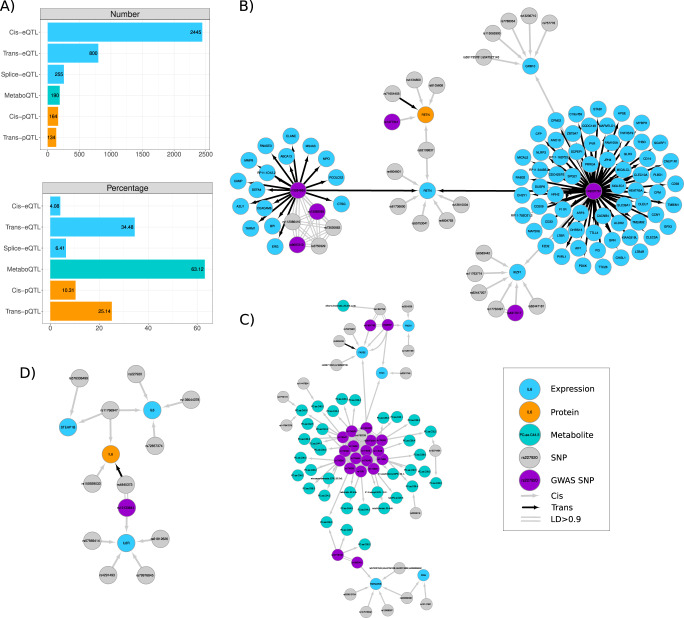
We evaluate the shared genetic regulation of mRNA molecules, proteins and metabolites derived from whole blood from 3029 human donors. We find abundant allelic heterogeneity, where multiple variants regulate a particular molecular phenotype, and pleiotropy, where a single variant associates with multiple molecular phenotypes over multiple genomic regions. The highest proportion of share genetic regulation is detected between gene expression and proteins (66.6%), with a further median shared genetic associations across 49 different tissues of 78.3% and 62.4% between plasma proteins and gene expression. We represent the genetic and molecular associations in networks including 2828 known GWAS variants, showing that GWAS variants are more often connected to gene expression in trans than other molecular phenotypes in the network. Our work provides a roadmap to understanding molecular networks and deriving the underlying mechanism of action of GWAS variants using different molecular phenotypes in an accessible tissue.
© 2023. Springer Nature Limited.
Conflict of interest statement
S.B. has ownerships in Intomics A/S, Hoba Therapeutics Aps, Novo Nordisk A/S, Lundbeck A/S, and managing board memberships in Proscion A/S and Intomics A/S. As of June 2019, M.I.M is an employee of Genentech and a holder of Roche stock. E.P. has received honoraria from Sanofi and Lilly. The other authors declare no competing interests. E.T.D. is currently an employee of GSK. The work presented in this manuscript was performed before he joined GSK. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
