

HDAC11 inhibition triggers bimodal thermogenic pathways to circumvent adipocyte catecholamine resistance
- PMID: 37607030
- PMCID: PMC10541202
- DOI: 10.1172/JCI168192
HDAC11 inhibition triggers bimodal thermogenic pathways to circumvent adipocyte catecholamine resistance
Abstract
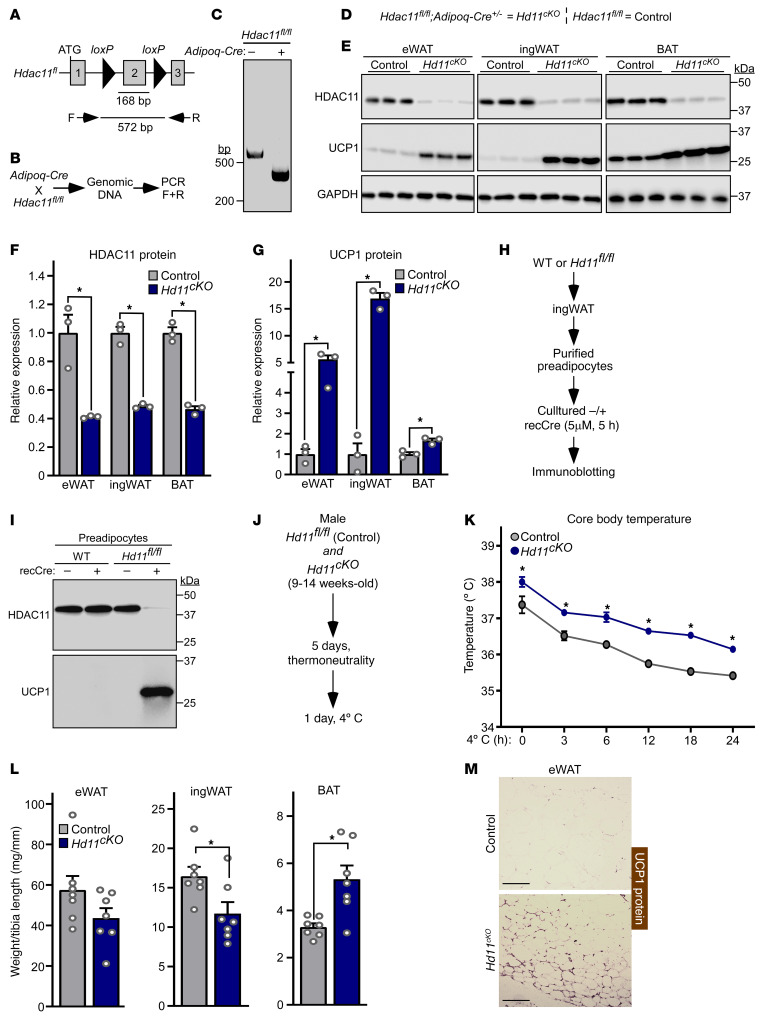
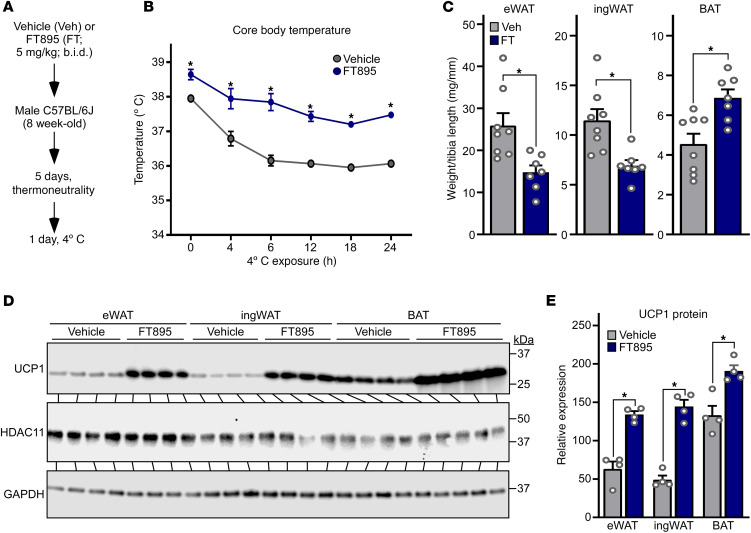
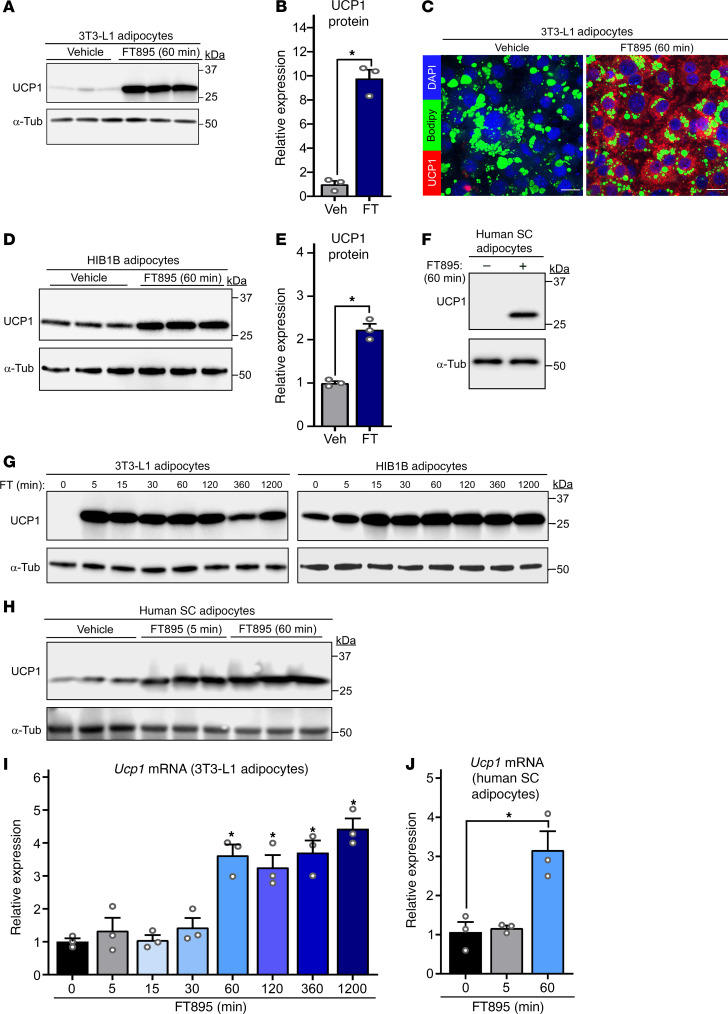
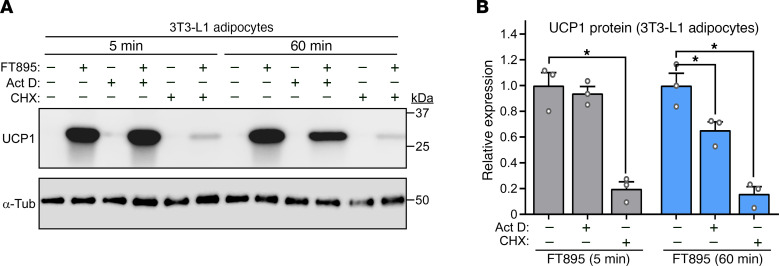
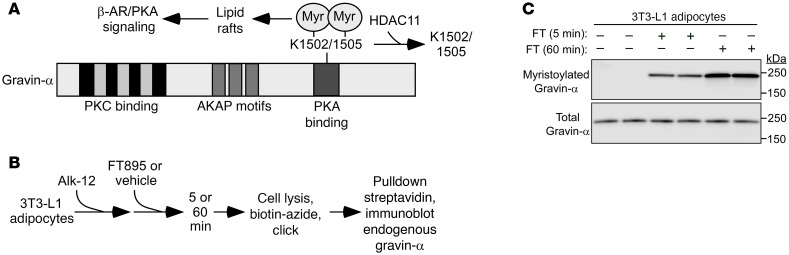
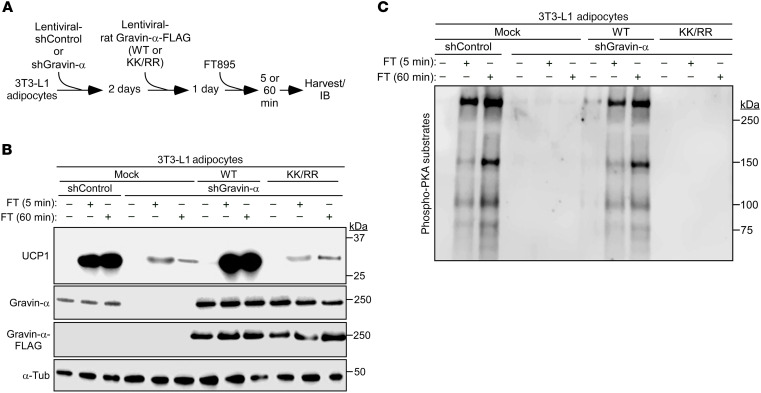
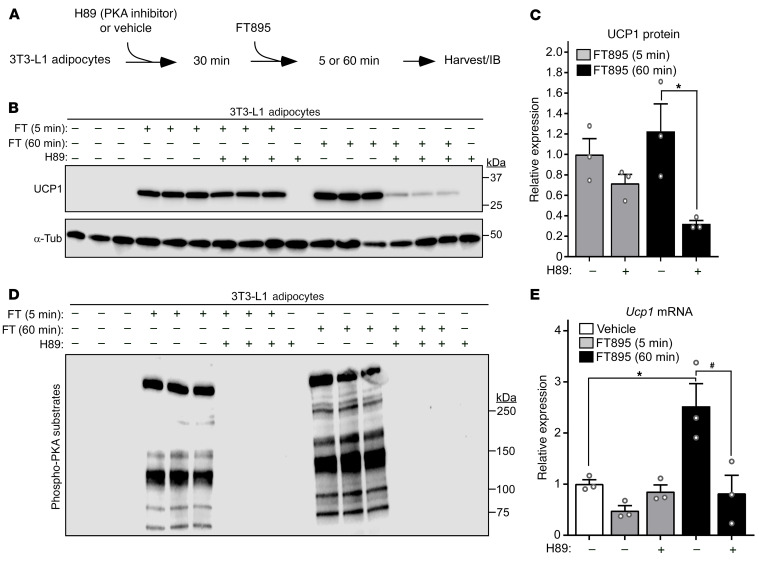
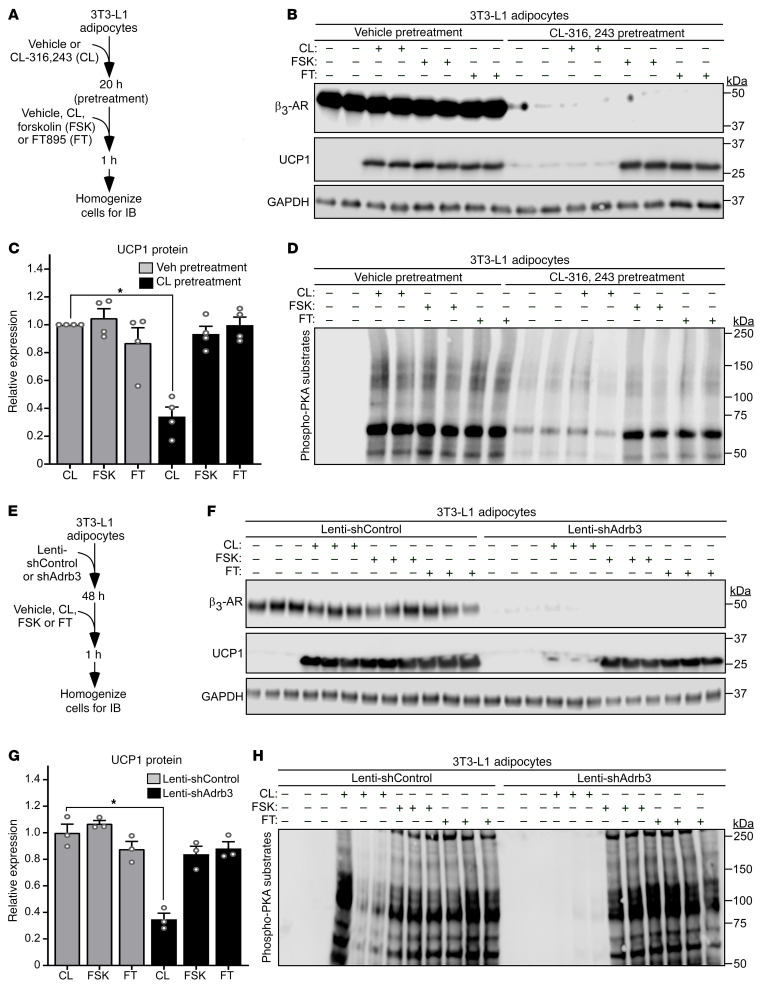
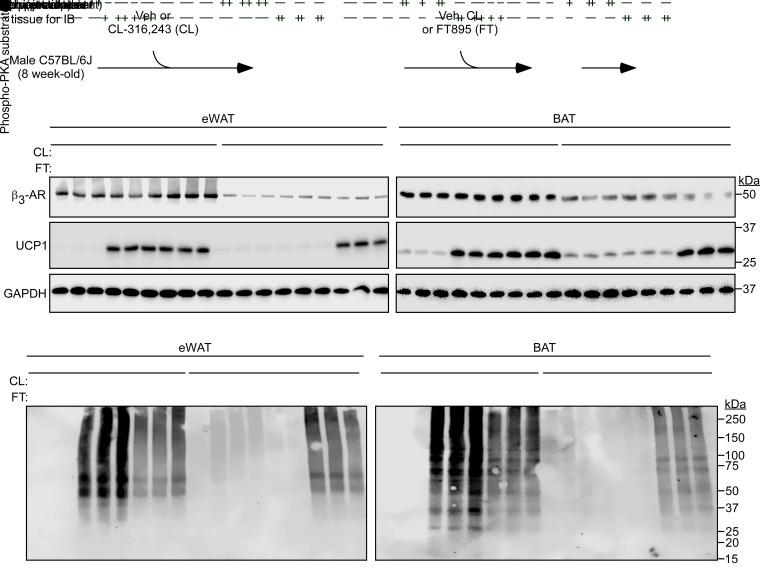
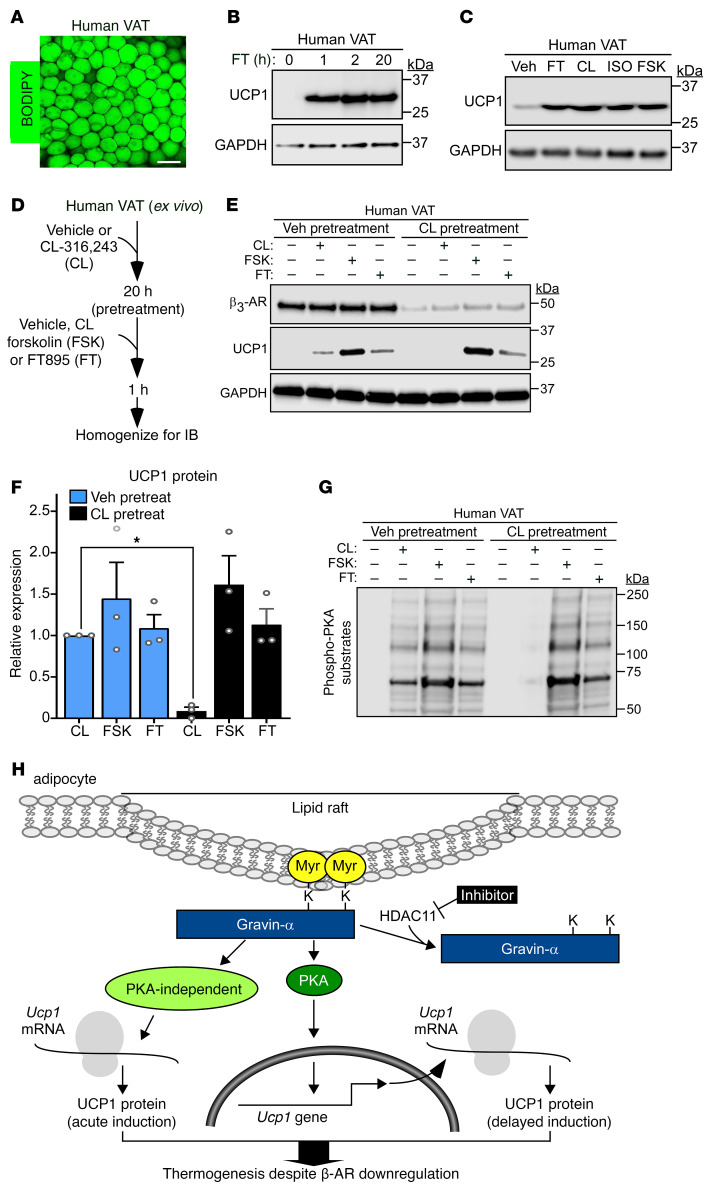
Stimulation of adipocyte β-adrenergic receptors (β-ARs) induces expression of uncoupling protein 1 (UCP1), promoting nonshivering thermogenesis. Association of β-ARs with a lysine-myristoylated form of A kinase-anchoring protein 12 (AKAP12, also known as gravin-α) is required for downstream signaling that culminates in UCP1 induction. Conversely, demyristoylation of gravin-α by histone deacetylase 11 (HDAC11) suppresses this pathway. Whether inhibition of HDAC11 in adipocytes is sufficient to drive UCP1 expression independently of β-ARs is not known. Here, we demonstrate that adipocyte-specific deletion of HDAC11 in mice leads to robust induction of UCP1 in adipose tissue (AT), resulting in increased body temperature. These effects are mimicked by treating mice in vivo or human AT ex vivo with an HDAC11-selective inhibitor, FT895. FT895 triggers biphasic, gravin-α myristoylation-dependent induction of UCP1 protein expression, with a noncanonical acute response that is posttranscriptional and independent of protein kinase A (PKA), and a delayed response requiring PKA activity and new Ucp1 mRNA synthesis. Remarkably, HDAC11 inhibition promotes UCP1 expression even in models of adipocyte catecholamine resistance where β-AR signaling is blocked. These findings define cell-autonomous, multimodal roles for HDAC11 as a suppressor of thermogenesis, and highlight the potential of inhibiting HDAC11 to therapeutically alter AT phenotype independently of β-AR stimulation.
Keywords: Adipose tissue; G protein–coupled receptors; Metabolism.
Conflict of interest statement
Figures
Update of
-
HDAC11 inhibition triggers bimodal thermogenic pathways to circumvent adipocyte catecholamine resistance.bioRxiv [Preprint]. 2023 Mar 30:2023.03.29.534830. doi: 10.1101/2023.03.29.534830. bioRxiv. 2023. Update in: J Clin Invest. 2023 Oct 2;133(19):e168192. doi: 10.1172/JCI168192. PMID: 37034582 Free PMC article. Updated. Preprint.
Similar articles
-
HDAC11 inhibition triggers bimodal thermogenic pathways to circumvent adipocyte catecholamine resistance.bioRxiv [Preprint]. 2023 Mar 30:2023.03.29.534830. doi: 10.1101/2023.03.29.534830. bioRxiv. 2023. Update in: J Clin Invest. 2023 Oct 2;133(19):e168192. doi: 10.1172/JCI168192. PMID: 37034582 Free PMC article. Updated. Preprint.
-
Reversible lysine fatty acylation of an anchoring protein mediates adipocyte adrenergic signaling.Proc Natl Acad Sci U S A. 2022 Feb 15;119(7):e2119678119. doi: 10.1073/pnas.2119678119. Proc Natl Acad Sci U S A. 2022. PMID: 35149557 Free PMC article.
-
Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis.Nat Med. 2017 May;23(5):623-630. doi: 10.1038/nm.4316. Epub 2017 Apr 17. Nat Med. 2017. PMID: 28414329 Free PMC article.
-
Meaningful respirometric measurements of UCP1-mediated thermogenesis.Biochimie. 2017 Mar;134:56-61. doi: 10.1016/j.biochi.2016.12.005. Epub 2016 Dec 14. Biochimie. 2017. PMID: 27986537 Review.
-
UCP1, the mitochondrial uncoupling protein of brown adipocyte: A personal contribution and a historical perspective.Biochimie. 2017 Mar;134:3-8. doi: 10.1016/j.biochi.2016.10.018. Epub 2016 Dec 2. Biochimie. 2017. PMID: 27916641 Review.
Cited by
-
Pathological mechanisms of cold and mechanical stress in modulating cancer progression.Hum Cell. 2024 May;37(3):593-606. doi: 10.1007/s13577-024-01049-y. Epub 2024 Mar 27. Hum Cell. 2024. PMID: 38538930 Review.
-
Gravi-D peptide disrupts HDAC11 association with an AKAP to stimulate adipocyte thermogenic signaling.J Clin Invest. 2024 May 1;134(9):e177726. doi: 10.1172/JCI177726. J Clin Invest. 2024. PMID: 38690735 Free PMC article. No abstract available.
-
Targeting Histone Deacetylase 11 with a Highly Selective Inhibitor for the Treatment of MASLD.Adv Sci (Weinh). 2025 Apr;12(15):e2412903. doi: 10.1002/advs.202412903. Epub 2025 Feb 20. Adv Sci (Weinh). 2025. PMID: 39976110 Free PMC article.
-
HDAC11 promotes renal fibrosis by induing partial epithelial-mesenchymal transition and G2/M phase arrest in renal epithelial cells.Res Sq [Preprint]. 2025 May 6:rs.3.rs-6523050. doi: 10.21203/rs.3.rs-6523050/v1. Res Sq. 2025. PMID: 40386380 Free PMC article. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
