

RNF4 and USP7 cooperate in ubiquitin-regulated steps of DNA replication
- PMID: 37607592
- PMCID: PMC10444366
- DOI: 10.1098/rsob.230068
RNF4 and USP7 cooperate in ubiquitin-regulated steps of DNA replication
Abstract
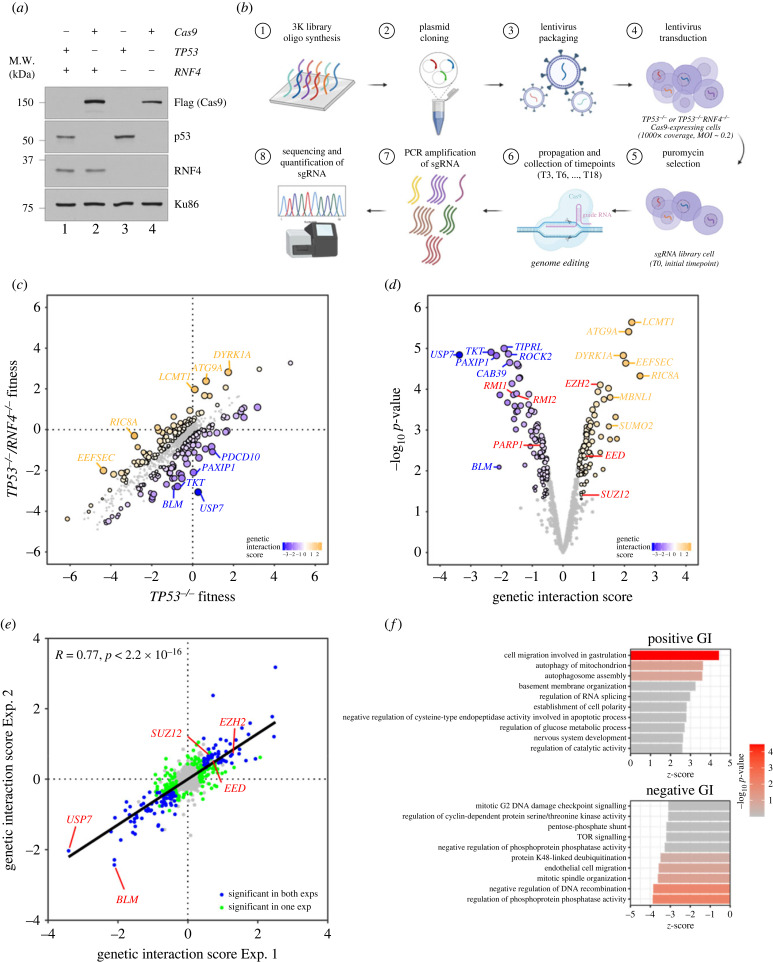
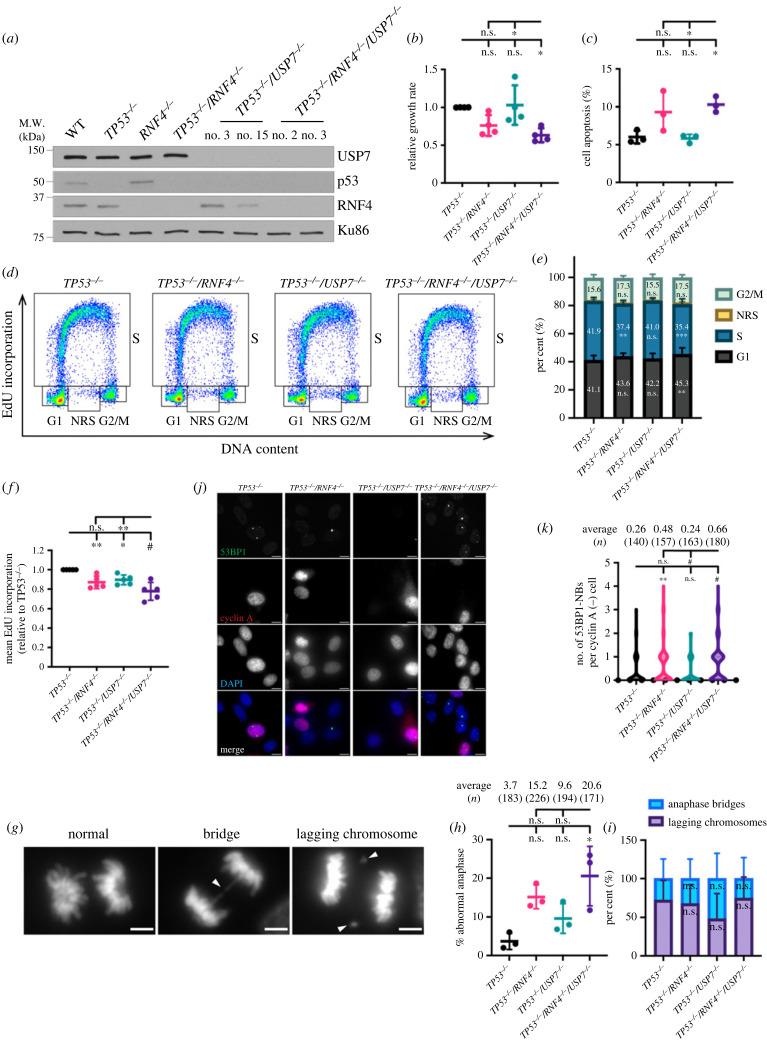
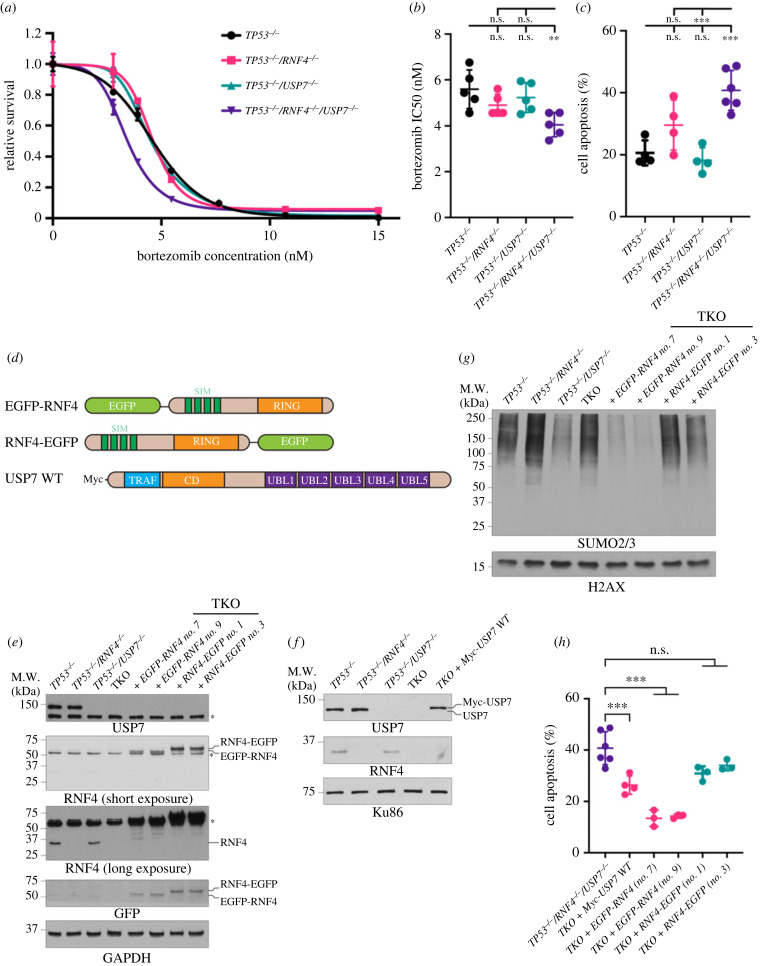
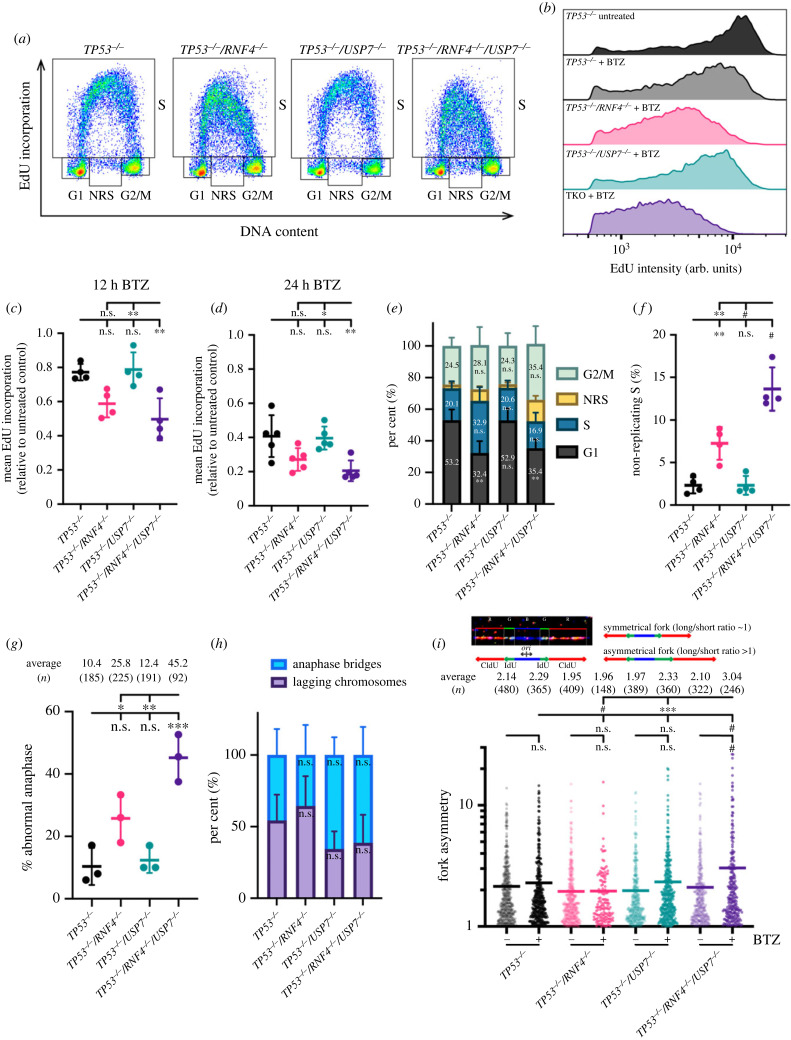
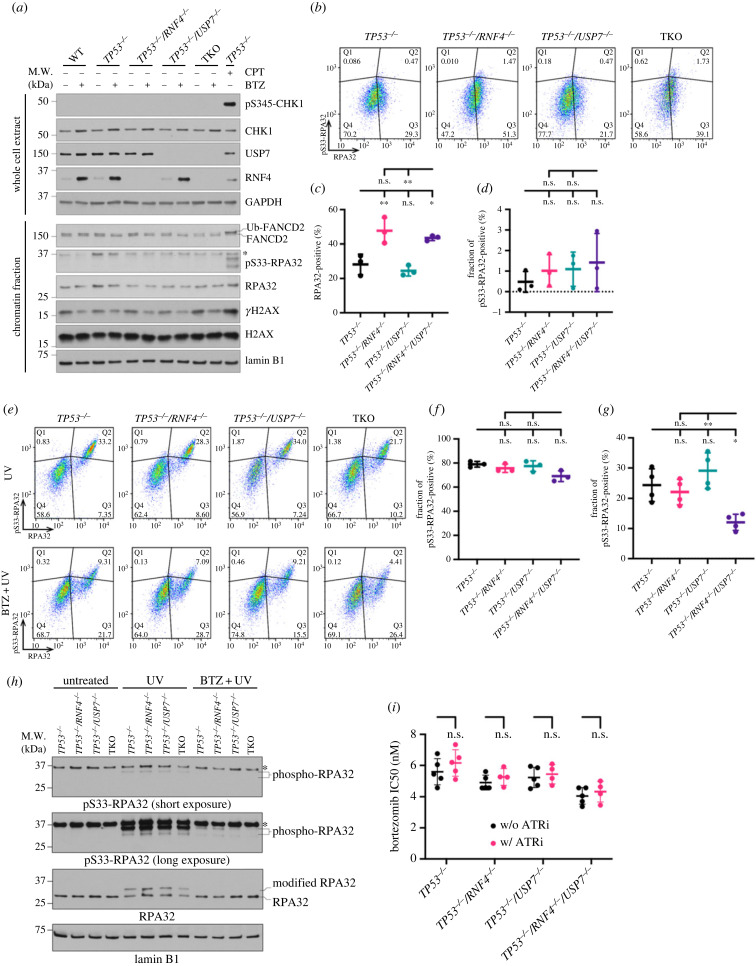
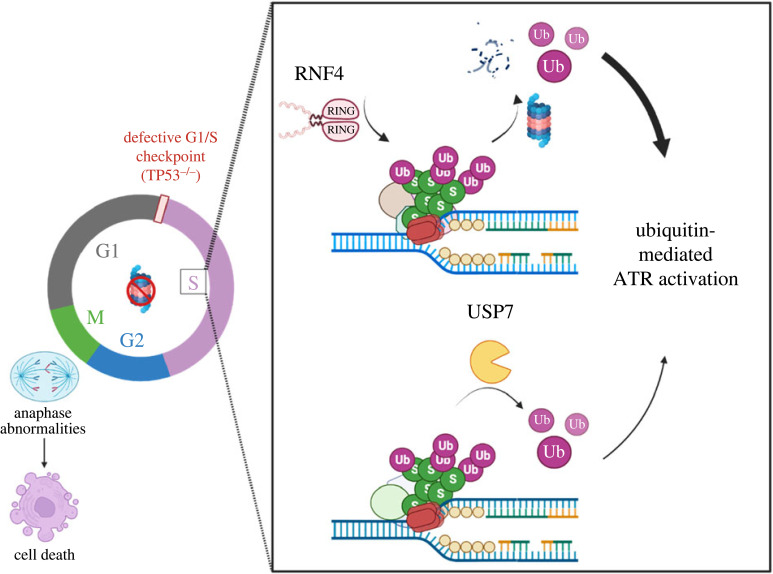
DNA replication requires precise regulation achieved through post-translational modifications, including ubiquitination and SUMOylation. These modifications are linked by the SUMO-targeted E3 ubiquitin ligases (STUbLs). Ring finger protein 4 (RNF4), one of only two mammalian STUbLs, participates in double-strand break repair and resolving DNA-protein cross-links. However, its role in DNA replication has been poorly understood. Using CRISPR/Cas9 genetic screens, we discovered an unexpected dependency of RNF4 mutants on ubiquitin specific peptidase 7 (USP7) for survival in TP53-null retinal pigment epithelial cells. TP53-/-/RNF4-/-/USP7-/- triple knockout (TKO) cells displayed defects in DNA replication that cause genomic instability. These defects were exacerbated by the proteasome inhibitor bortezomib, which limited the nuclear ubiquitin pool. A shortage of free ubiquitin suppressed the ataxia telangiectasia and Rad3-related (ATR)-mediated checkpoint response, leading to increased cell death. In conclusion, RNF4 and USP7 work cooperatively to sustain a functional level of nuclear ubiquitin to maintain the integrity of the genome.
Keywords: RNF4; STUbL; SUMO; USP7; genome stability; ubiquitin.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
E1B-55K-Mediated Regulation of RNF4 SUMO-Targeted Ubiquitin Ligase Promotes Human Adenovirus Gene Expression.J Virol. 2018 Jun 13;92(13):e00164-18. doi: 10.1128/JVI.00164-18. Print 2018 Jul 1. J Virol. 2018. PMID: 29695423 Free PMC article.
-
Methods to analyze STUbL activity.Methods Enzymol. 2019;618:257-280. doi: 10.1016/bs.mie.2018.11.005. Epub 2019 Jan 3. Methods Enzymol. 2019. PMID: 30850055
-
USP7 and VCPFAF1 define the SUMO/Ubiquitin landscape at the DNA replication fork.Cell Rep. 2021 Oct 12;37(2):109819. doi: 10.1016/j.celrep.2021.109819. Cell Rep. 2021. PMID: 34644576 Free PMC article.
-
SUMO-Targeted Ubiquitin Ligases and Their Functions in Maintaining Genome Stability.Int J Mol Sci. 2021 May 20;22(10):5391. doi: 10.3390/ijms22105391. Int J Mol Sci. 2021. PMID: 34065507 Free PMC article. Review.
-
RNF4-A Paradigm for SUMOylation-Mediated Ubiquitination.Proteomics. 2019 Nov;19(21-22):e1900185. doi: 10.1002/pmic.201900185. Epub 2019 Oct 29. Proteomics. 2019. PMID: 31566917 Review.
Cited by
-
RNF4 prevents genomic instability caused by chronic DNA under-replication.DNA Repair (Amst). 2024 Mar;135:103646. doi: 10.1016/j.dnarep.2024.103646. Epub 2024 Feb 7. DNA Repair (Amst). 2024. PMID: 38340377 Free PMC article.
-
The Role and Mechanism of Deubiquitinase USP7 in Tumor-Associated Inflammation.Biomedicines. 2024 Nov 29;12(12):2734. doi: 10.3390/biomedicines12122734. Biomedicines. 2024. PMID: 39767641 Free PMC article. Review.
-
SUMO and the DNA damage response.Biochem Soc Trans. 2024 Apr 24;52(2):773-792. doi: 10.1042/BST20230862. Biochem Soc Trans. 2024. PMID: 38629643 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
