

Demethylase-independent roles of LSD1 in regulating enhancers and cell fate transition
- PMID: 37607921
- PMCID: PMC10444793
- DOI: 10.1038/s41467-023-40606-1
Demethylase-independent roles of LSD1 in regulating enhancers and cell fate transition
Abstract
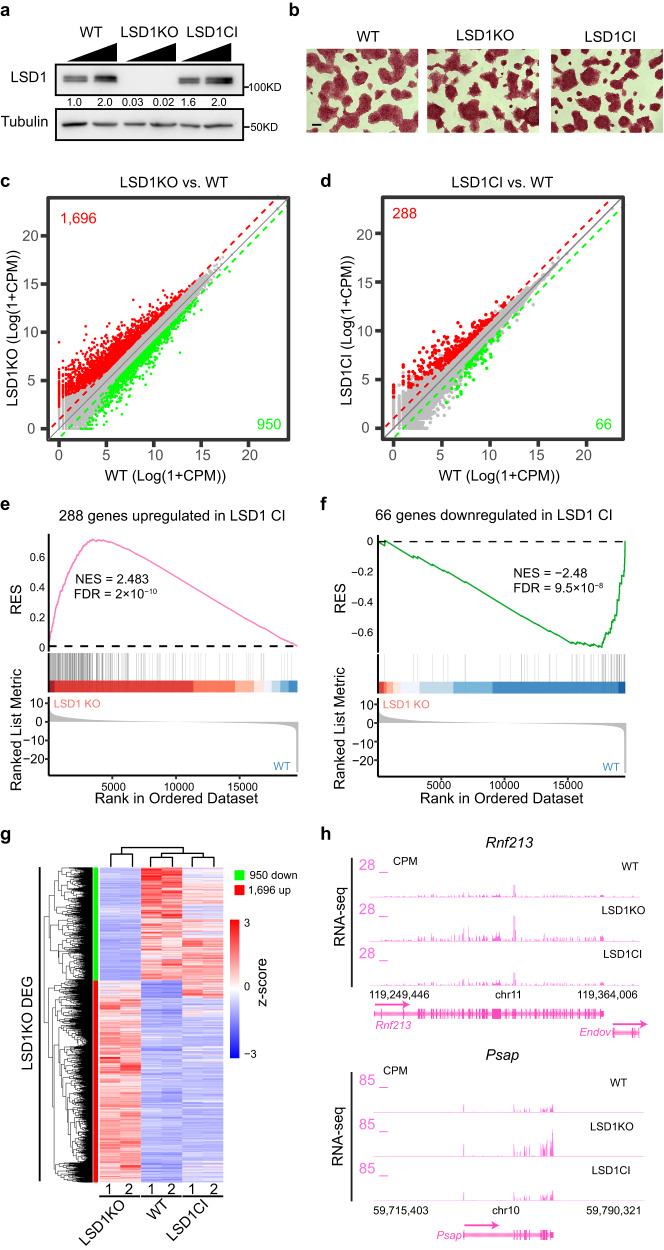
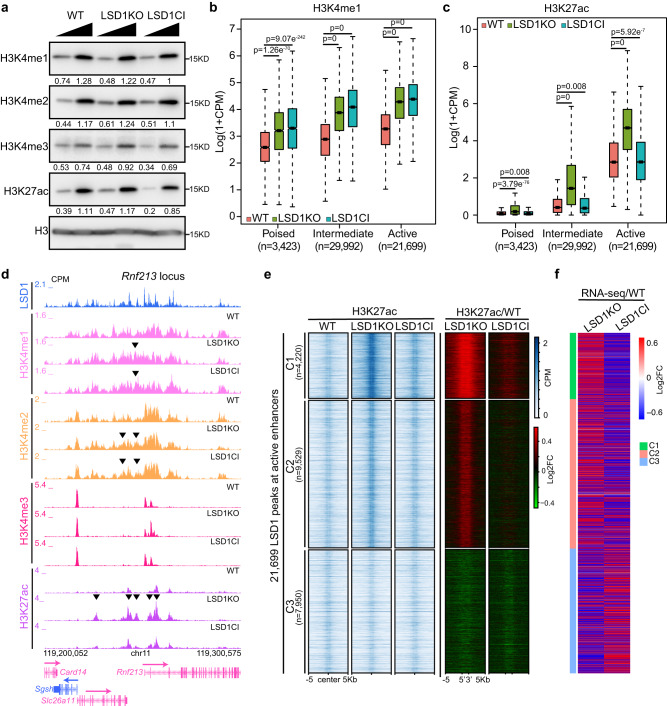
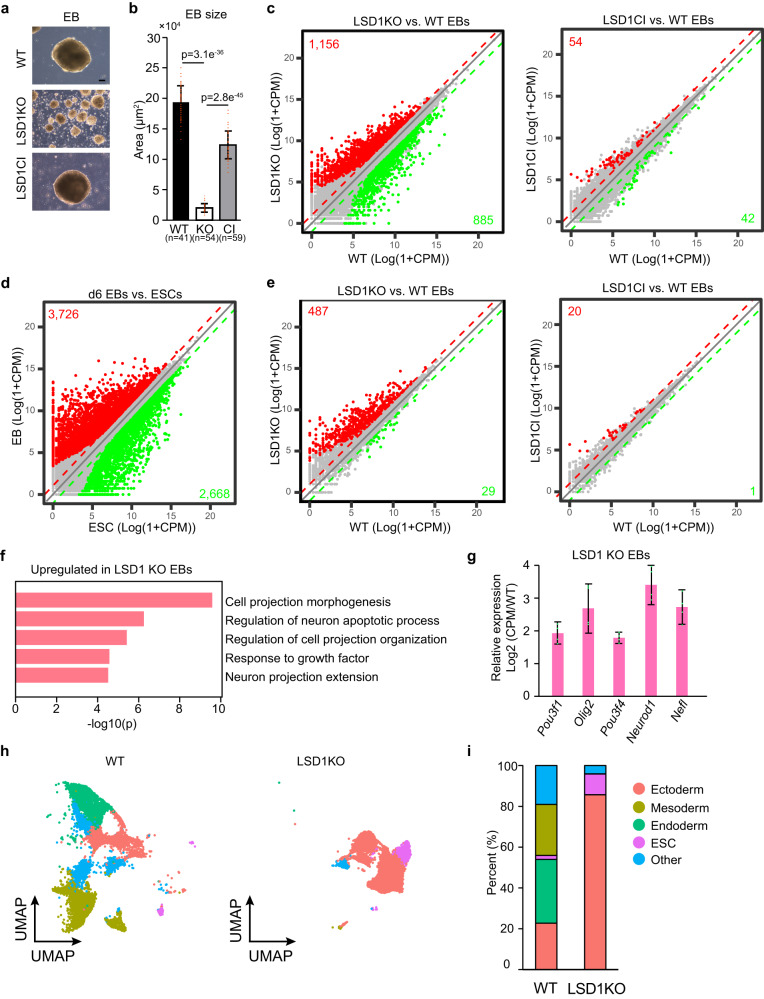
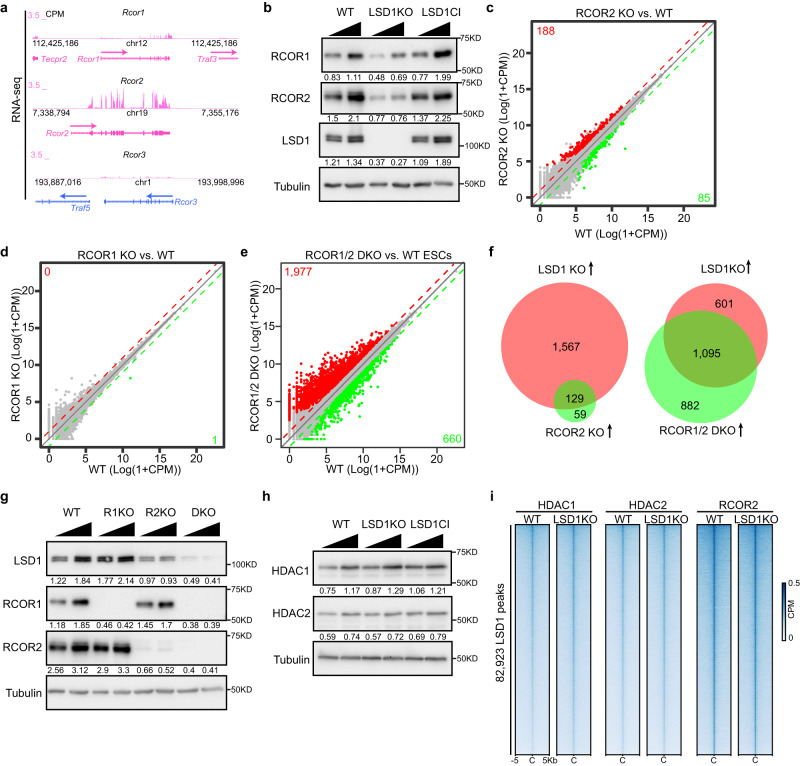
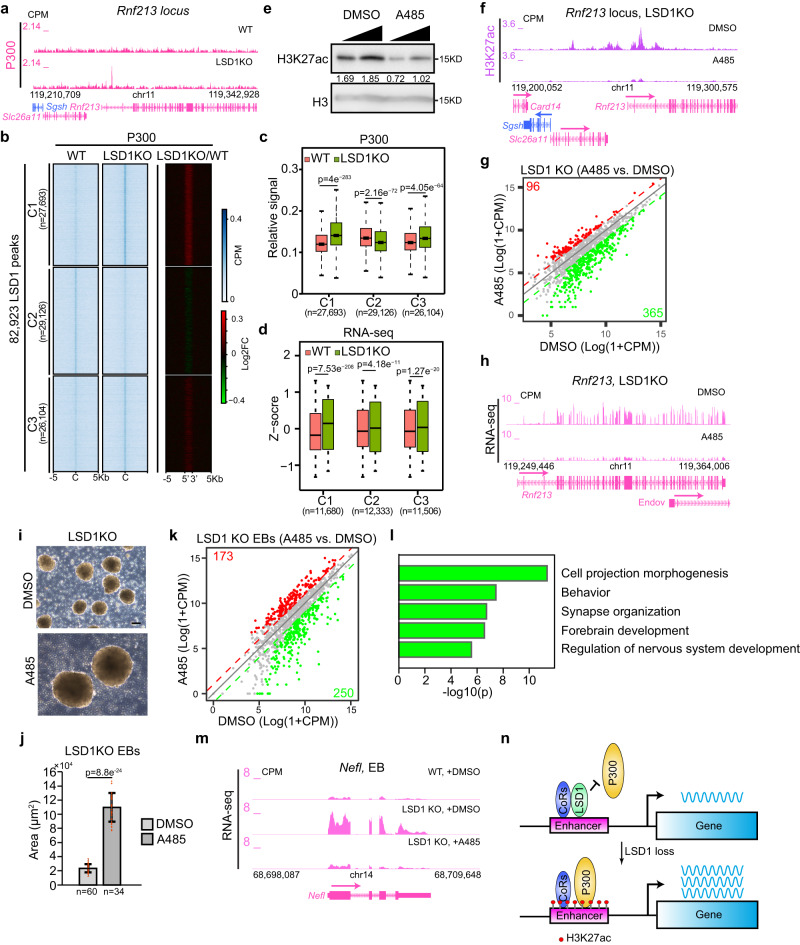
The major enhancer regulator lysine-specific histone demethylase 1A (LSD1) is required for mammalian embryogenesis and is implicated in human congenital diseases and multiple types of cancer; however, the underlying mechanisms remain enigmatic. Here, we dissect the role of LSD1 and its demethylase activity in gene regulation and cell fate transition. Surprisingly, the catalytic inactivation of LSD1 has a mild impact on gene expression and cellular differentiation whereas the loss of LSD1 protein de-represses enhancers globally and impairs cell fate transition. LSD1 deletion increases H3K27ac levels and P300 occupancy at LSD1-targeted enhancers. The gain of H3K27ac catalyzed by P300/CBP, not the loss of CoREST complex components from chromatin, contributes to the transcription de-repression of LSD1 targets and differentiation defects caused by LSD1 loss. Together, our study demonstrates a demethylase-independent role of LSD1 in regulating enhancers and cell fate transition, providing insight into treating diseases driven by LSD1 mutations and misregulation.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
