

Convergent somatic evolution commences in utero in a germline ribosomopathy
- PMID: 37608017
- PMCID: PMC10444798
- DOI: 10.1038/s41467-023-40896-5
Convergent somatic evolution commences in utero in a germline ribosomopathy
Abstract
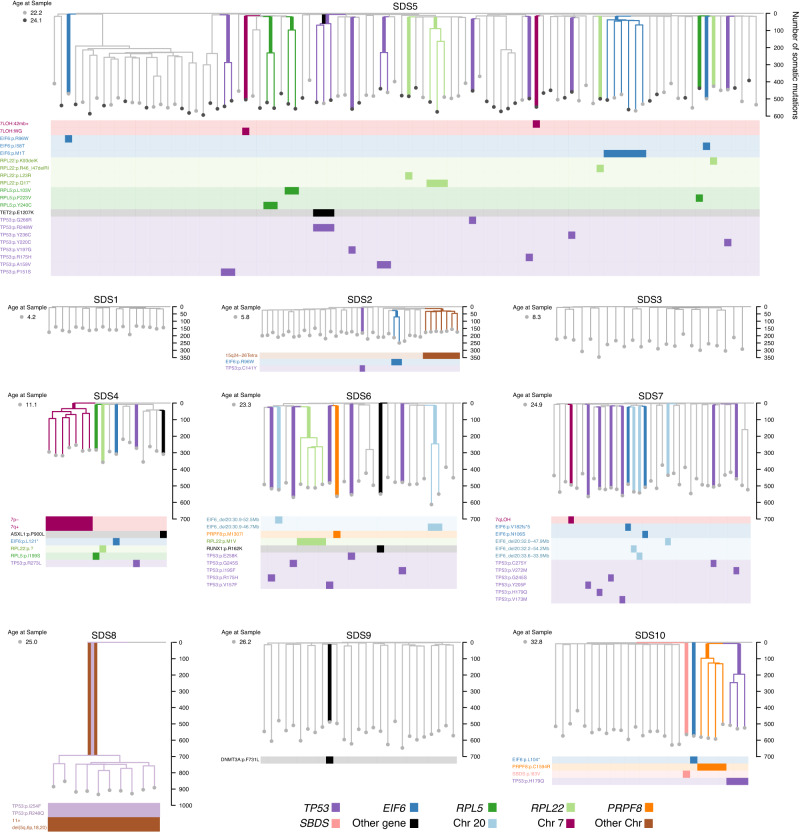
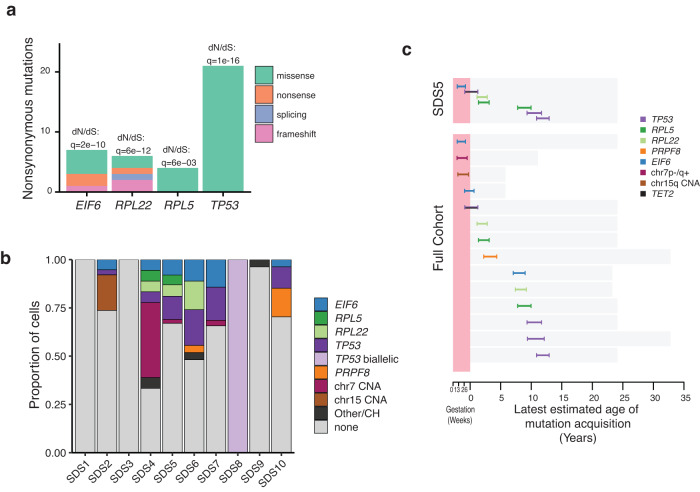
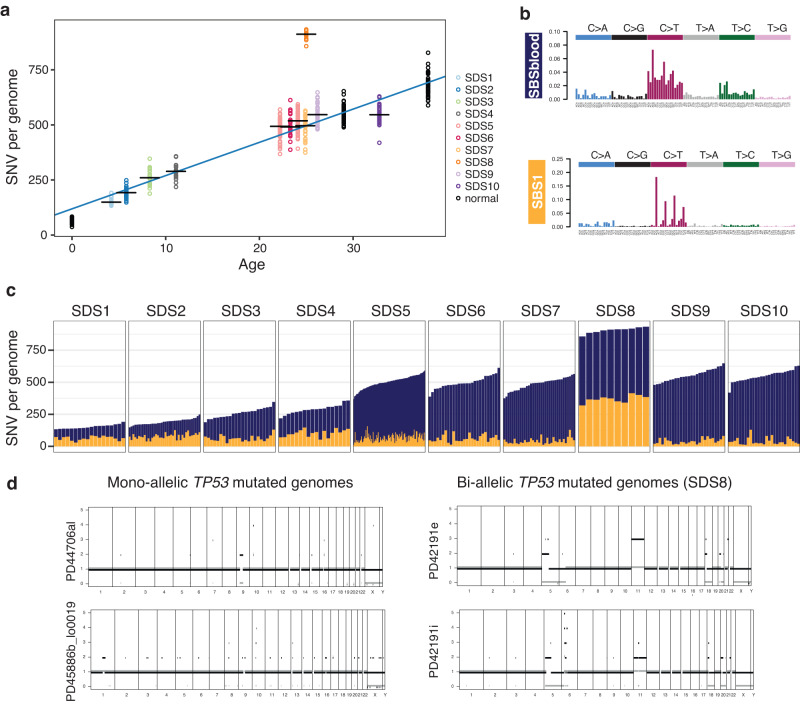
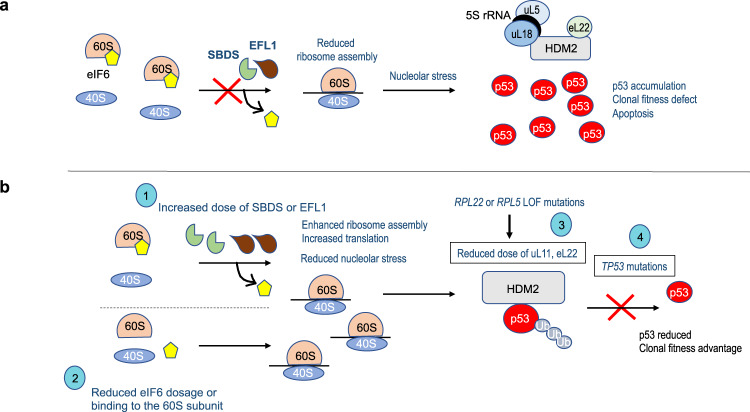
Clonal tracking of cells using somatic mutations permits exploration of clonal dynamics in human disease. Here, we perform whole genome sequencing of 323 haematopoietic colonies from 10 individuals with the inherited ribosomopathy Shwachman-Diamond syndrome to reconstruct haematopoietic phylogenies. In ~30% of colonies, we identify mutually exclusive mutations in TP53, EIF6, RPL5, RPL22, PRPF8, plus chromosome 7 and 15 aberrations that increase SBDS and EFL1 gene dosage, respectively. Target gene mutations commence in utero, resulting in a profusion of clonal expansions, with only a few haematopoietic stem cell lineages (mean 8, range 1-24) contributing ~50% of haematopoietic colonies across 8 individuals (range 4-100% clonality) by young adulthood. Rapid clonal expansion during disease transformation is associated with biallelic TP53 mutations and increased mutation burden. Our study highlights how convergent somatic mutation of the p53-dependent nucleolar surveillance pathway offsets the deleterious effects of germline ribosomopathy but increases opportunity for TP53-mutated cancer evolution.
© 2023. Springer Nature Limited.
Conflict of interest statement
A.J.W. and S.T. are consultants for SDS Therapeutics. P.J.C. is a cofounder and shareholder of FL86 Inc. The remaining authors declare no competing interests.
Figures


References
-
- Williams N, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature. 2022;602:162–168. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
