

BioConvert: a comprehensive format converter for life sciences
- PMID: 37608802
- PMCID: PMC10440784
- DOI: 10.1093/nargab/lqad074
BioConvert: a comprehensive format converter for life sciences
Abstract
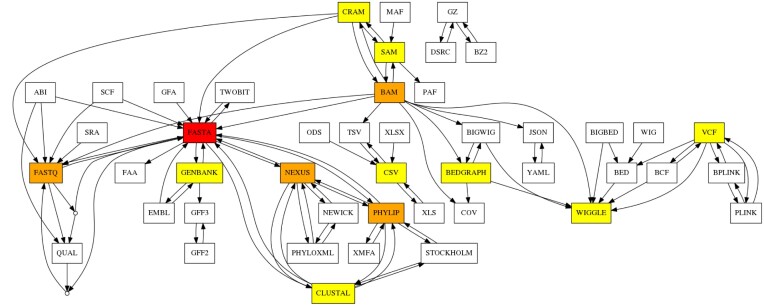
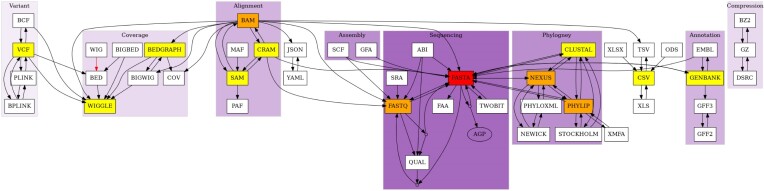
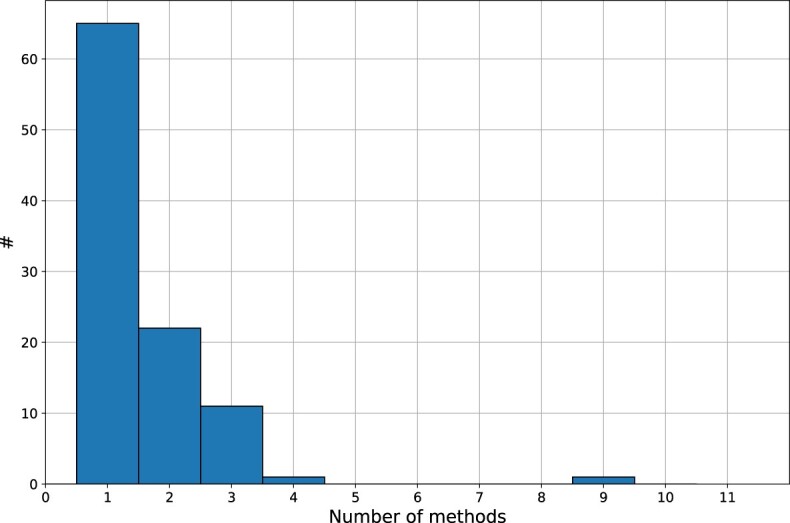
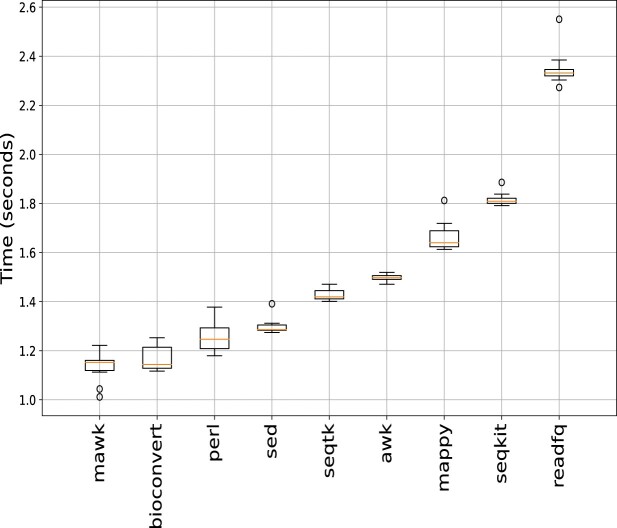
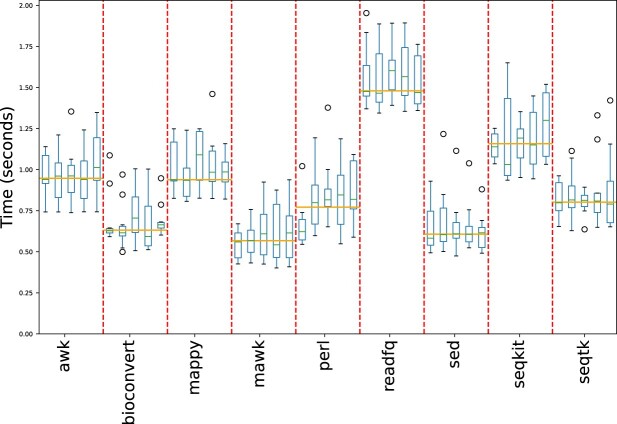
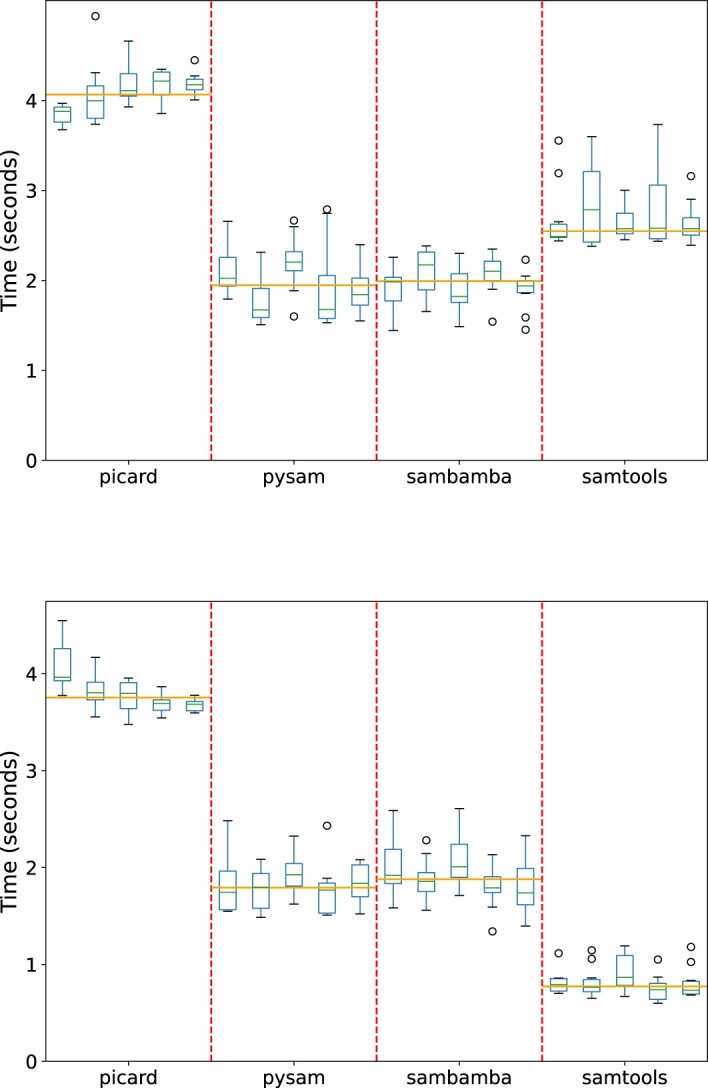
Bioinformatics is a field known for the numerous standards and formats that have been developed over the years. This plethora of formats, sometimes complementary, and often redundant, poses many challenges to bioinformatics data analysts. They constantly need to find the best tool to convert their data into the suitable format, which is often a complex, technical and time consuming task. Moreover, these small yet important tasks are often difficult to make reproducible. To overcome these difficulties, we initiated BioConvert, a collaborative project to facilitate the conversion of life science data from one format to another. BioConvert aggregates existing software within a single framework and complemented them with original code when needed. It provides a common interface to make the user experience more streamlined instead of having to learn tens of them. Currently, BioConvert supports about 50 formats and 100 direct conversions in areas such as alignment, sequencing, phylogeny, and variant calling. In addition to being useful for end-users, BioConvert can also be utilized by developers as a universal benchmarking framework for evaluating and comparing numerous conversion tools. Additionally, we provide a web server implementing an online user-friendly interface to BioConvert, hence allowing direct use for the community.
© The Author(s) 2023. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures






References
-
- Stein L. Creating a bioinformatics nation. Nature. 2002; 417:119–120. - PubMed
-
- Andrews S., Krueger F., Segonds-Pichon A., Biggins L., Krueger C., Wingett S.. FASTQC. A quality control tool for high throughput sequence data. 2010;
-
- Ewels P.A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M.U., Di Tommaso P., Nahnsen S.. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020; 38:276–278. - PubMed
LinkOut - more resources
Full Text Sources
