

This is a preprint.
The role of APOBEC3-induced mutations in the differential evolution of monkeypox virus
- PMID: 37608930
- PMCID: PMC10441442
The role of APOBEC3-induced mutations in the differential evolution of monkeypox virus
Update in
-
The role of APOBEC3-induced mutations in the differential evolution of monkeypox virus.Virus Evol. 2023 Oct 5;9(2):vead058. doi: 10.1093/ve/vead058. eCollection 2023. Virus Evol. 2023. PMID: 37841642 Free PMC article.
Abstract
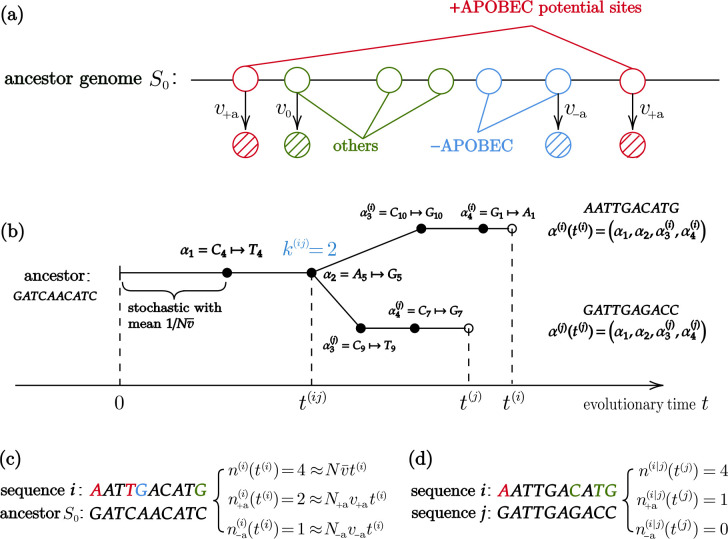
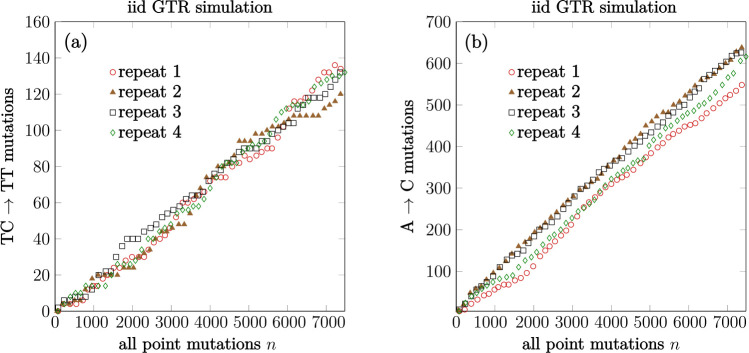
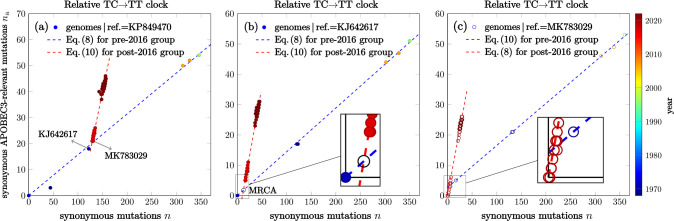
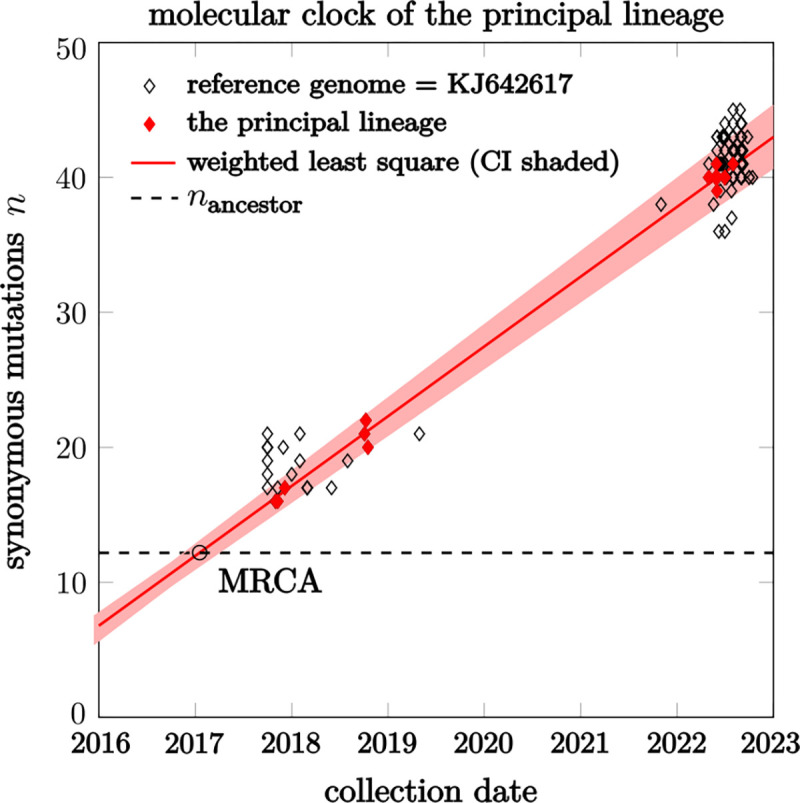
Recent studies show that newly sampled monkeypox virus (MPXV) genomes exhibit mutations consistent with Apolipoprotein B mRNA Editing Catalytic Polypeptide-like3 (APOBEC3)-mediated editing, compared to MPXV genomes collected earlier. It is unclear whether these single nucleotide polymorphisms (SNPs) result from APOBEC3-induced editing or are a consequence of genetic drift within one or more MPXV animal reservoirs. We develop a simple method based on a generalization of the General-Time-Reversible (GTR) model to show that the observed SNPs are likely the result of APOBEC3-induced editing. The statistical features allow us to extract lineage information and estimate evolutionary events.
Conflict of interest statement
Disclosure and competing interests The authors have no competing interests.
Figures
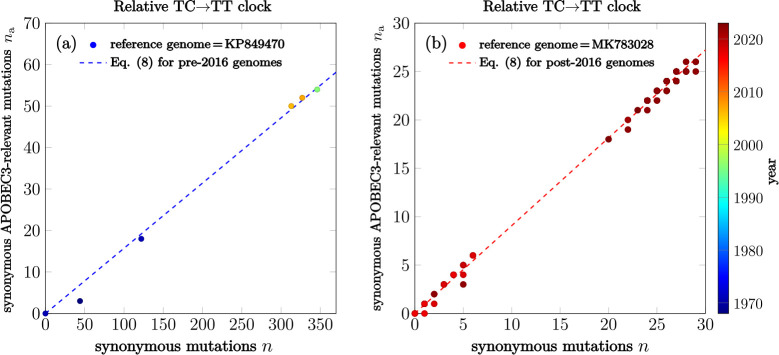
Similar articles
-
The role of APOBEC3-induced mutations in the differential evolution of monkeypox virus.Virus Evol. 2023 Oct 5;9(2):vead058. doi: 10.1093/ve/vead058. eCollection 2023. Virus Evol. 2023. PMID: 37841642 Free PMC article.
-
Sequencing of monkeypox virus from infected patients reveals viral genomes with APOBEC3-like editing, gene inactivation, and bacterial agents of skin superinfection.J Med Virol. 2023 Jun;95(6):e28799. doi: 10.1002/jmv.28799. J Med Virol. 2023. PMID: 37342884
-
Evolutionary potential of the monkeypox genome arising from interactions with human APOBEC3 enzymes.Virus Evol. 2023 Aug 2;9(2):vead047. doi: 10.1093/ve/vead047. eCollection 2023. Virus Evol. 2023. PMID: 37577211 Free PMC article.
-
Molecular Aspects of the Monkeypox Virus and their Impact on the Virus's Change in Epidemiology.P R Health Sci J. 2024 Sep;43(3):111-118. P R Health Sci J. 2024. PMID: 39269761 Review.
-
Differences in pathogenicity among the mpox virus clades: impact on drug discovery and vaccine development.Trends Pharmacol Sci. 2023 Oct;44(10):719-739. doi: 10.1016/j.tips.2023.08.003. Epub 2023 Sep 4. Trends Pharmacol Sci. 2023. PMID: 37673695 Review.
References
-
- Áine O’Toole and Andrew Rambaut. An APOBEC3 molecular clock to estimate the date of emergence of hMPXV. https://virological.org/t/an-apobec3-molecular-clock-to-estimate-the-dat..., 2022. (Accessed on 10/16/2022).
-
- Áine O’Toole and Andrew Rambaut. Initial observations about putative APOBEC3 deaminase editing driving short-term evolution of MPXV since 2017. https://virological.org/t/initial-observations-about-putative-apobec3-de..., 2022. (Accessed on 10/16/2022).
-
- Áine O’Toole and Andrew Rambaut. Update to observations about putative APOBEC3 deaminase editing in the light of new genomes from USA. https://virological.org/t/update-to-observations-about-putative-apobec3-..., 2022. (Accessed on 10/16/2022).