

This is a preprint.
Using deep long-read RNAseq in Alzheimer's disease brain to assess medical relevance of RNA isoform diversity
- PMID: 37609156
- PMCID: PMC10441303
- DOI: 10.1101/2023.08.06.552162
Using deep long-read RNAseq in Alzheimer's disease brain to assess medical relevance of RNA isoform diversity
Update in
-
Mapping medically relevant RNA isoform diversity in the aged human frontal cortex with deep long-read RNA-seq.Nat Biotechnol. 2025 Apr;43(4):635-646. doi: 10.1038/s41587-024-02245-9. Epub 2024 May 22. Nat Biotechnol. 2025. PMID: 38778214 Free PMC article.
Abstract
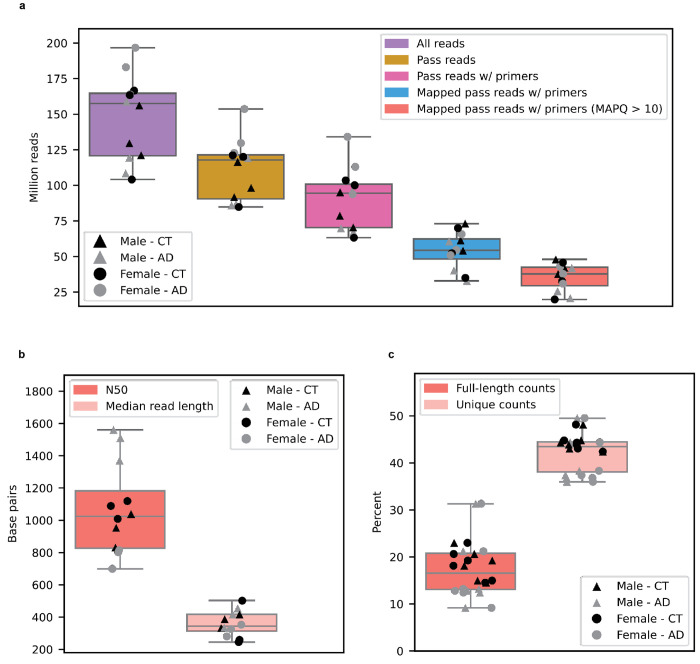
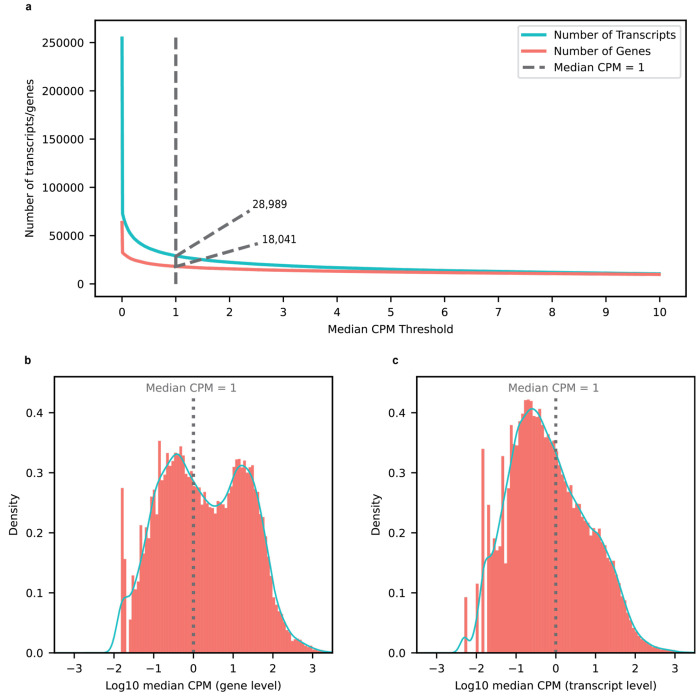
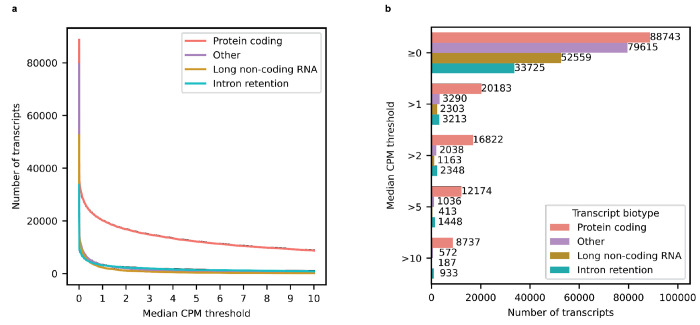
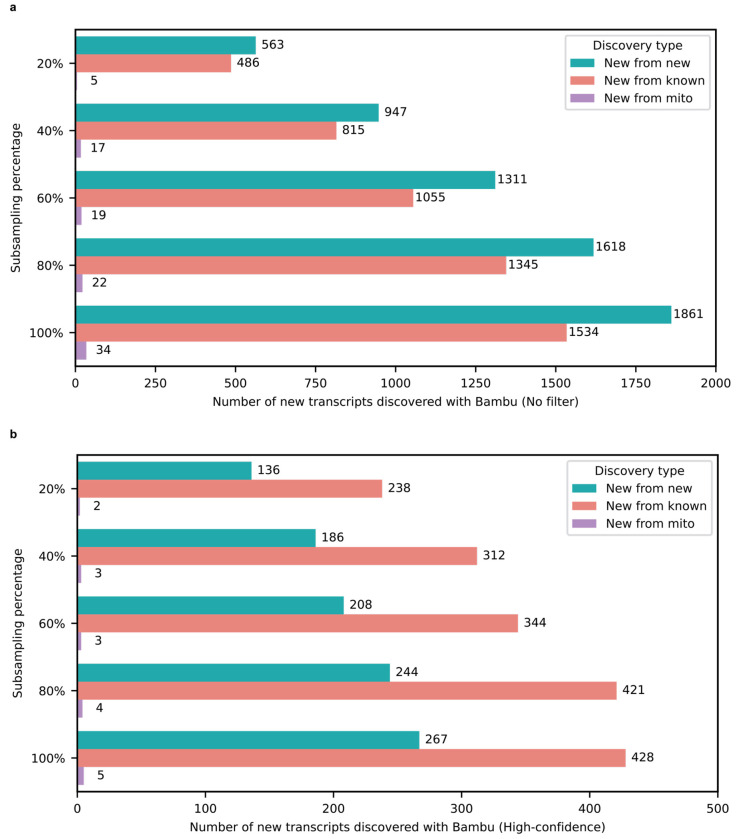
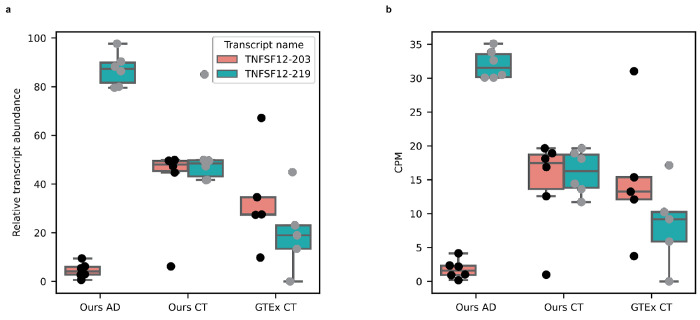
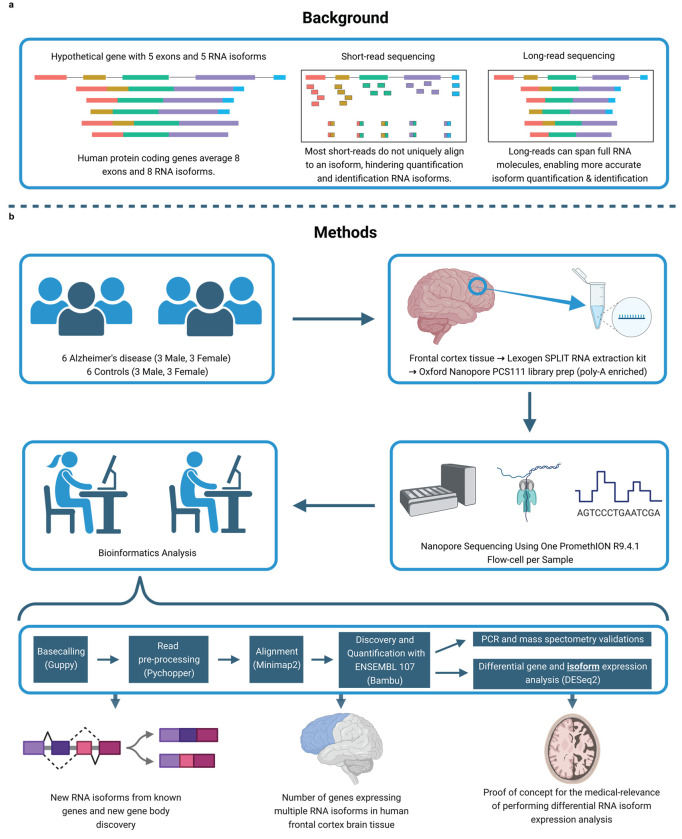
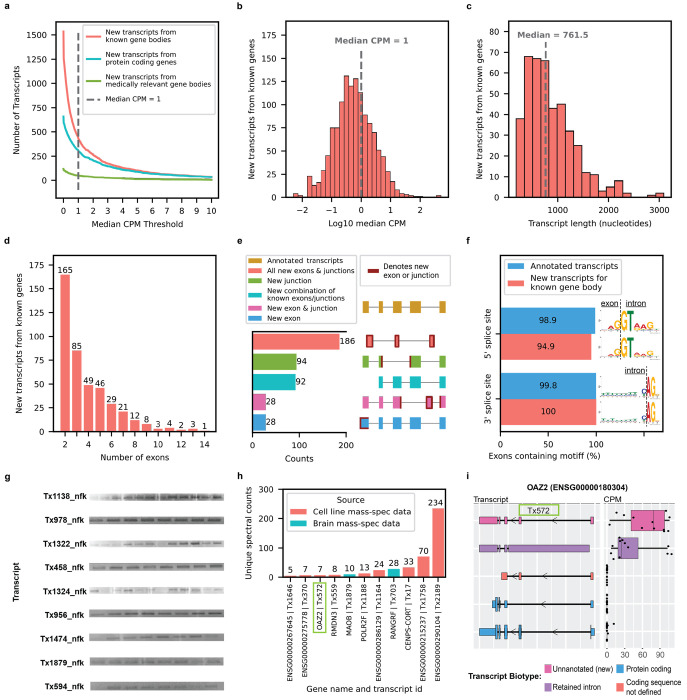
Due to alternative splicing, human protein-coding genes average over eight RNA isoforms, resulting in nearly four distinct protein coding sequences per gene. Long-read RNAseq (IsoSeq) enables more accurate quantification of isoforms, shedding light on their specific roles. To assess the medical relevance of measuring RNA isoform expression, we sequenced 12 aged human frontal cortices (6 Alzheimer's disease cases and 6 controls; 50% female) using one Oxford Nanopore PromethION flow cell per sample. Our study uncovered 53 new high-confidence RNA isoforms in medically relevant genes, including several where the new isoform was one of the most highly expressed for that gene. Specific examples include WDR4 (61%; microcephaly), MYL3 (44%; hypertrophic cardiomyopathy), and MTHFS (25%; major depression, schizophrenia, bipolar disorder). Other notable genes with new high-confidence isoforms include CPLX2 (10%; schizophrenia, epilepsy) and MAOB (9%; targeted for Parkinson's disease treatment). We identified 1,917 medically relevant genes expressing multiple isoforms in human frontal cortex, where 1,018 had multiple isoforms with different protein coding sequences, demonstrating the need to better understand how individual isoforms from a single gene body are involved in human health and disease, if at all. Exactly 98 of the 1,917 genes are implicated in brain-related diseases, including Alzheimer's disease genes such as APP (Aβ precursor protein; five), MAPT (tau protein; four), and BIN1 (eight). As proof of concept, we also found 99 differentially expressed RNA isoforms between Alzheimer's cases and controls, despite the genes themselves not exhibiting differential expression. Our findings highlight the significant knowledge gaps in RNA isoform diversity and their medical relevance. Deep long-read RNA sequencing will be necessary going forward to fully comprehend the medical relevance of individual isoforms for a "single" gene.
Keywords: Alzheimer’s disease; Human brain; Long reads; Medical relevance; Nanopore sequencing; RNA isoforms.
Conflict of interest statement
Competing interests The authors report no competing interests.
Figures









References
-
- Oberwinkler J., Lis A., Giehl K. M., Flockerzi V. & Philipp S. E. Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J. Biol. Chem. 280, 22540–22548 (2005). - PubMed
-
- Végran F. et al. Overexpression of caspase-3s splice variant in locally advanced breast carcinoma is associated with poor response to neoadjuvant chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 12, 5794–5800 (2006). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials