

Primary aldosteronism: molecular medicine meets public health
- PMID: 37612380
- PMCID: PMC7615304
- DOI: 10.1038/s41581-023-00753-6
Primary aldosteronism: molecular medicine meets public health
Abstract
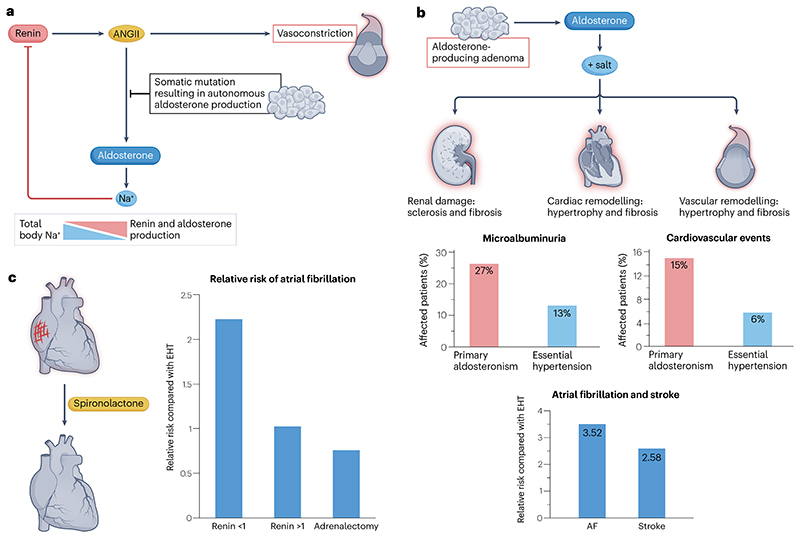
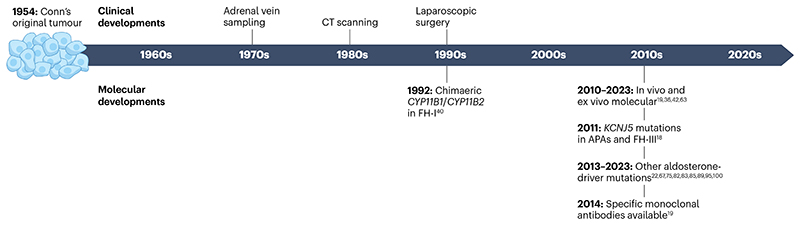
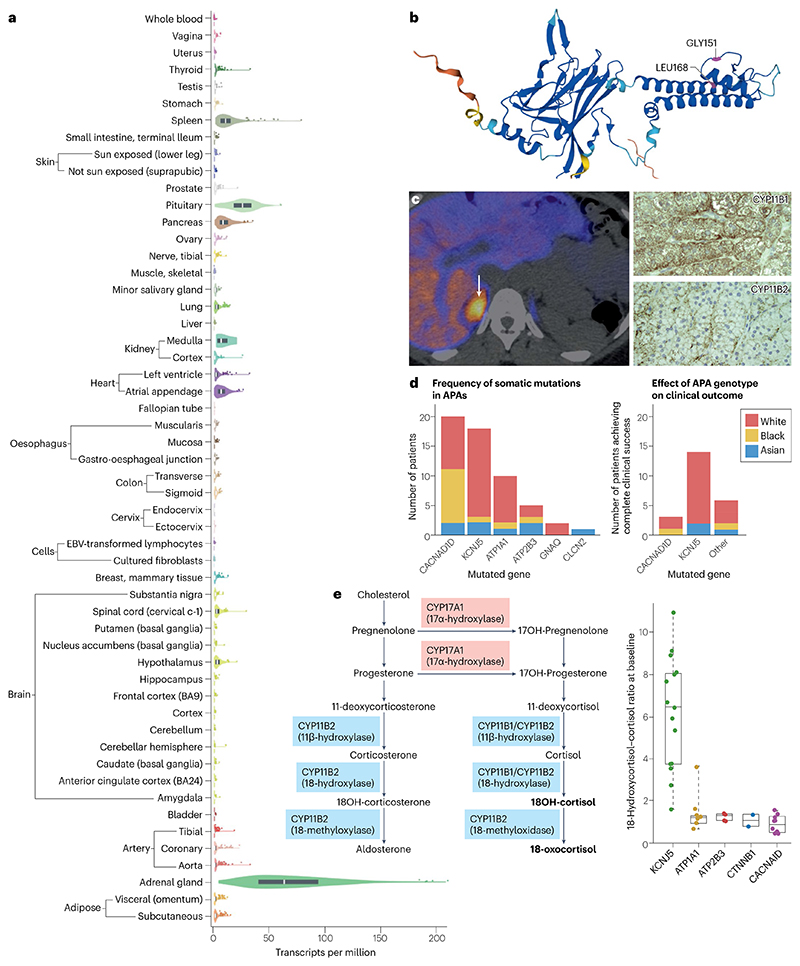
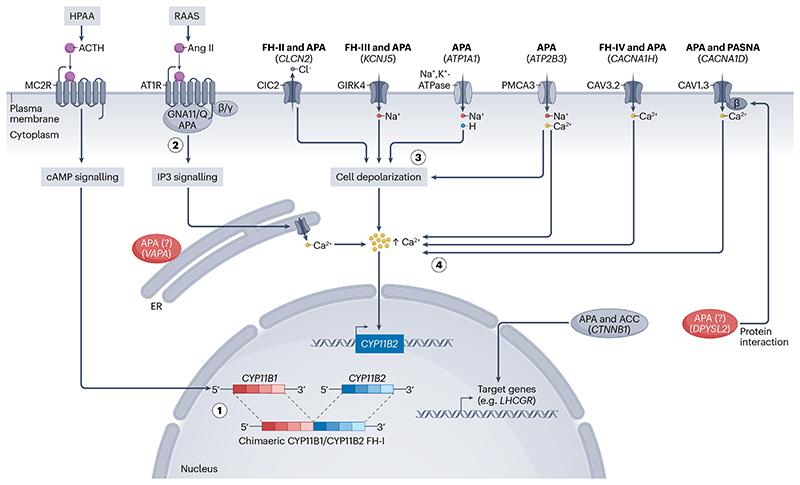
Primary aldosteronism is the most common single cause of hypertension and is potentially curable when only one adrenal gland is the culprit. The importance of primary aldosteronism to public health derives from its high prevalence but huge under-diagnosis (estimated to be <1% of all affected individuals), despite the consequences of poor blood pressure control by conventional therapy and enhanced cardiovascular risk. This state of affairs is attributable to the fact that the tools used for diagnosis or treatment are still those that originated in the 1970-1990s. Conversely, molecular discoveries have transformed our understanding of adrenal physiology and pathology. Many molecules and processes associated with constant adrenocortical renewal and interzonal metamorphosis also feature in aldosterone-producing adenomas and aldosterone-producing micronodules. The adrenal gland has one of the most significant rates of non-silent somatic mutations, with frequent selection of those driving autonomous aldosterone production, and distinct clinical presentations and outcomes for most genotypes. The disappearance of aldosterone synthesis and cells from most of the adult human zona glomerulosa is the likely driver of the mutational success that causes aldosterone-producing adenomas, but insights into the pathways that lead to constitutive aldosterone production and cell survival may open up opportunities for novel therapies.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Simpson SA, et al. [Isolation from the adrenals of a new crystalline hormone with especially high effectiveness on mineral metabolism] Experientia. 1953;9:333–335. - PubMed
-
- Conn JW, Louis LH. Primary aldosteronism, a new clinical entity. Ann Intern Med. 1956;44:1–15. - PubMed
-
- Andersen GS, Toftdahl DB, Lund JO, Strandgaard S, Nielsen PE. The incidence rate of phaeochromocytoma and Conn’s syndrome in Denmark, 1977-1981. J Hum Hypertens. 1988;2:187–189. - PubMed
-
- Grell GA, Hanchard B, Fletcher PR, Clarke WF, Williams W. Conn’s syndrome. Case report and a review of the syndrome of primary aldosteronism (SOPA) West Indian Med J. 1984;33:48–54. - PubMed
-
- Simpson F, Barnett A. Primary aldosteronism (Conn’s syndrome): report of a case, and an analysis of published cases. Med J Aust. 1961;1:729–735.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
