

Diversity and complexity of cell death: a historical review
- PMID: 37612413
- PMCID: PMC10474147
- DOI: 10.1038/s12276-023-01078-x
Diversity and complexity of cell death: a historical review
Erratum in
-
Author Correction: Diversity and complexity of cell death: a historical review.Exp Mol Med. 2023 Sep;55(9):2083. doi: 10.1038/s12276-023-01107-9. Exp Mol Med. 2023. PMID: 37731035 Free PMC article. No abstract available.
Abstract
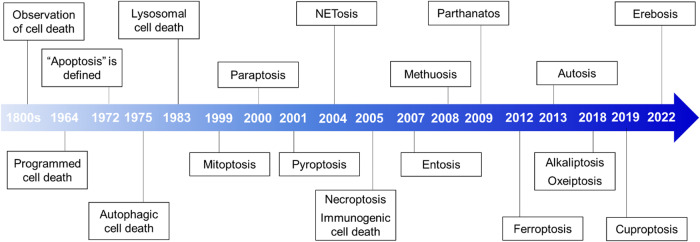
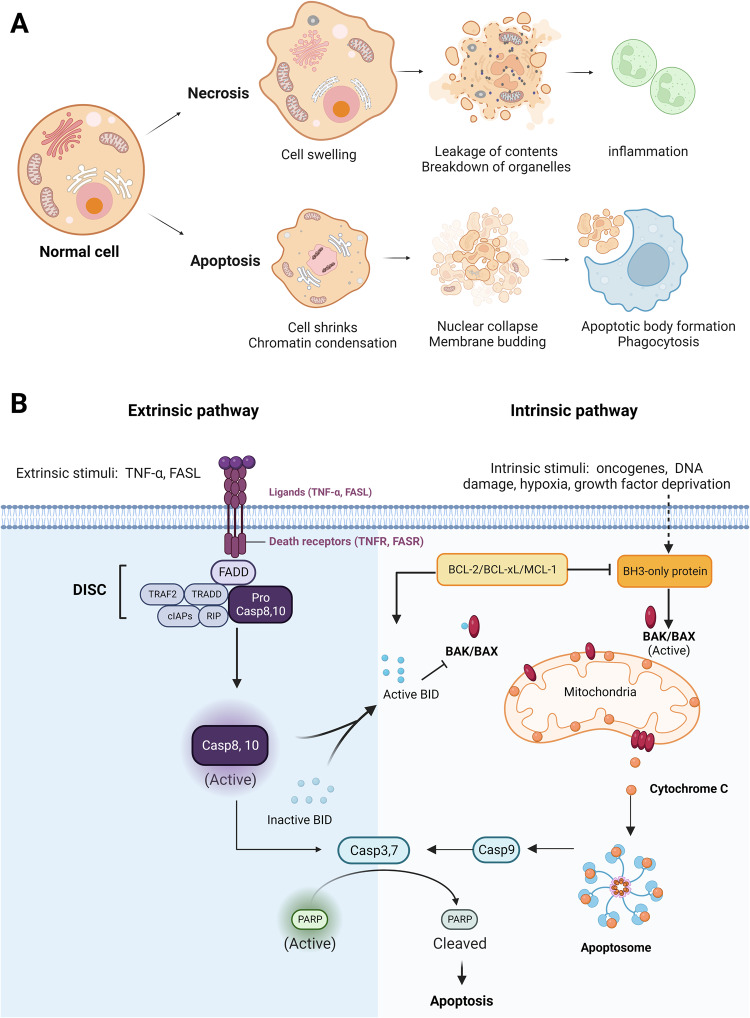
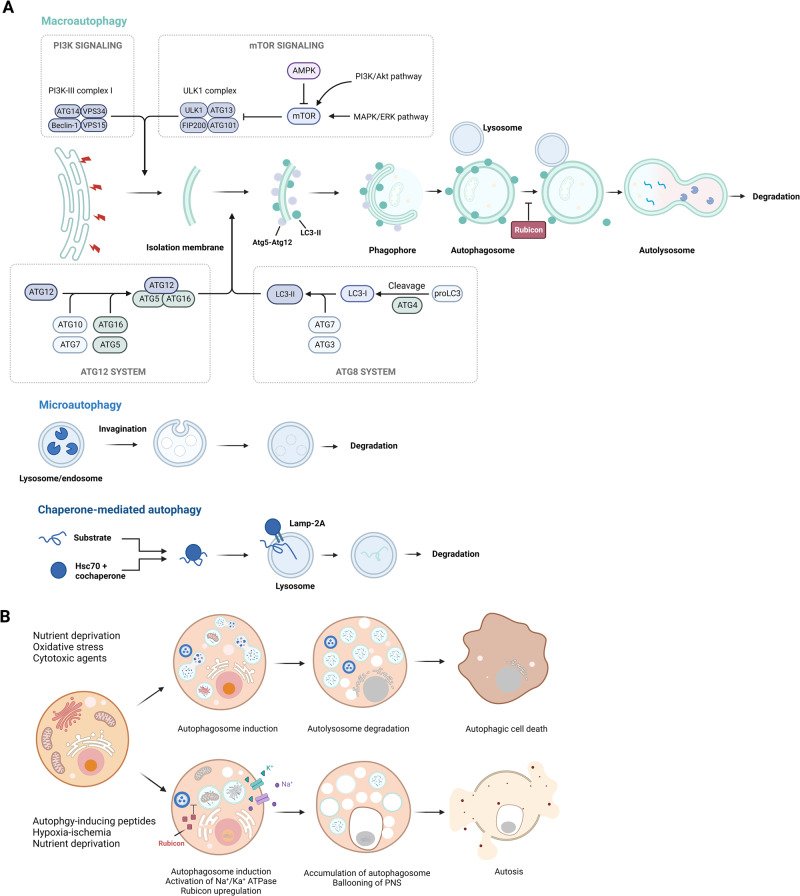
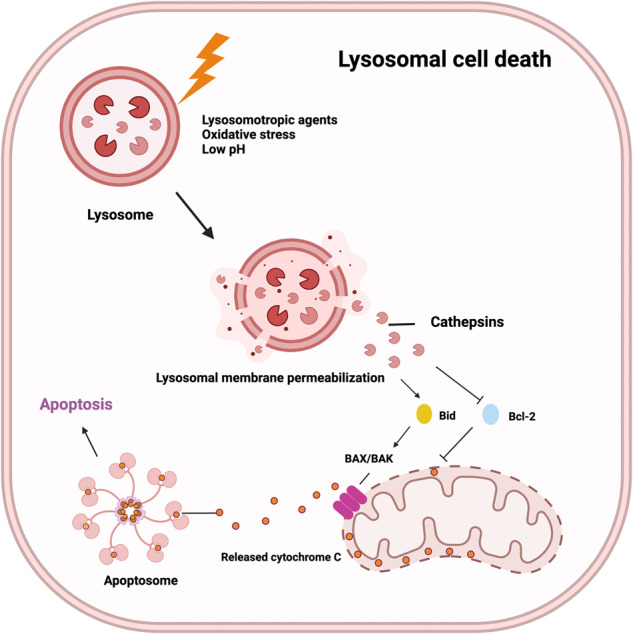
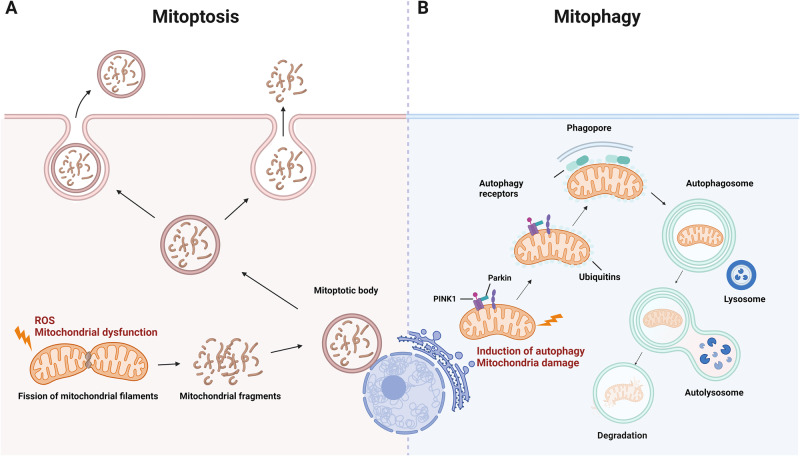
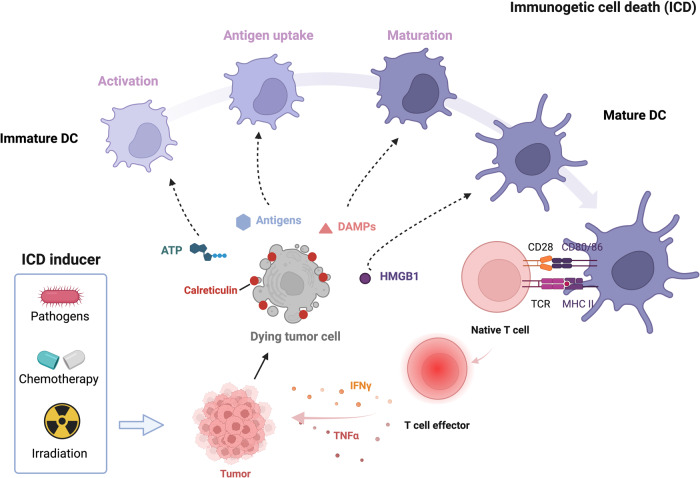
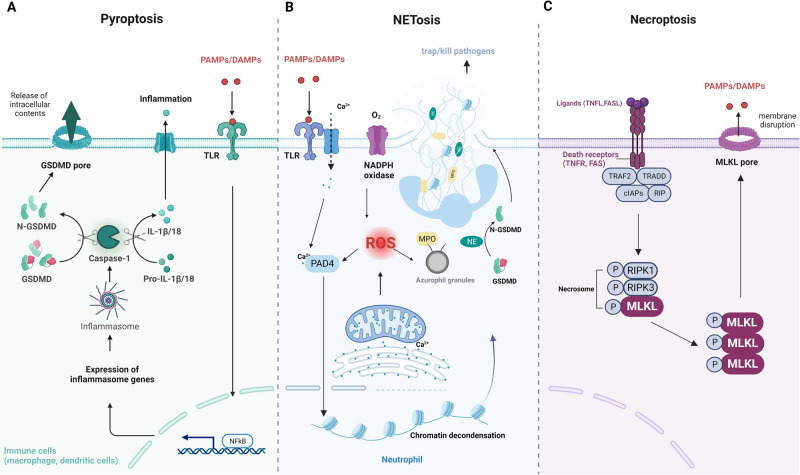
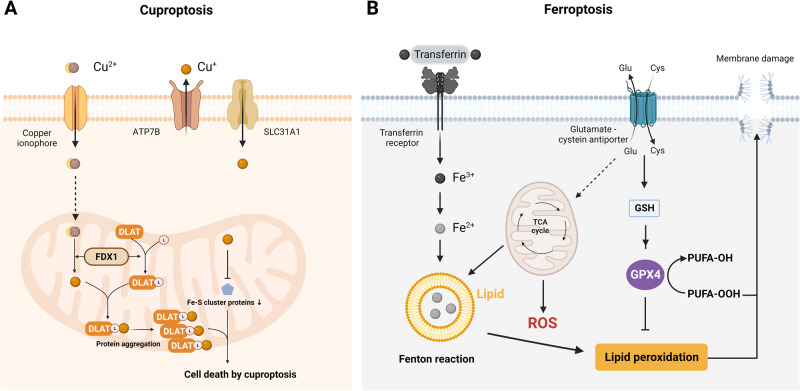
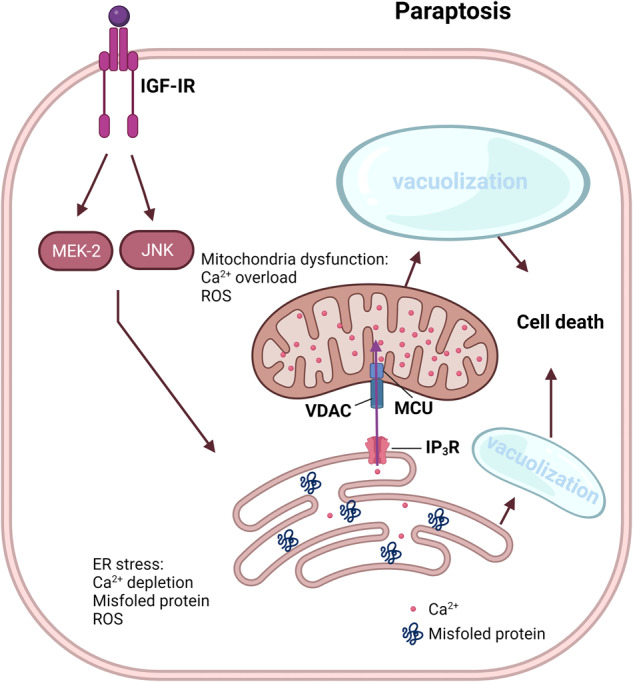
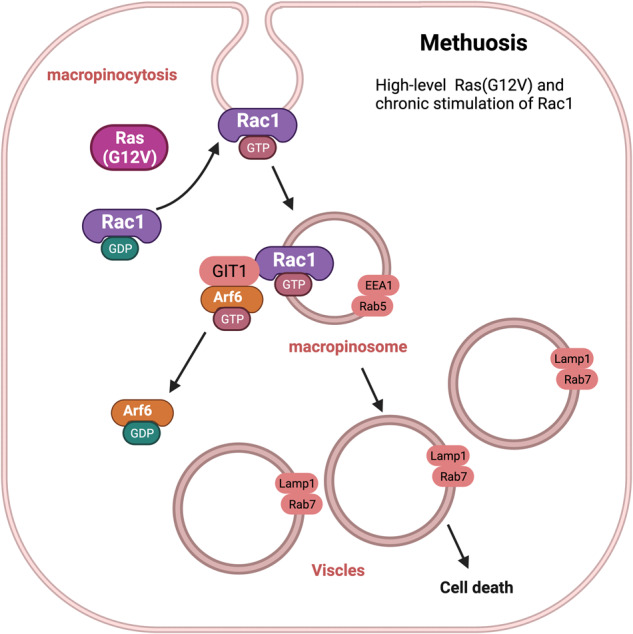
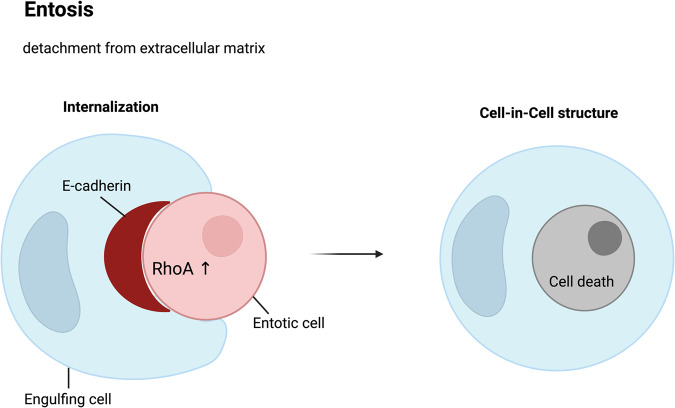
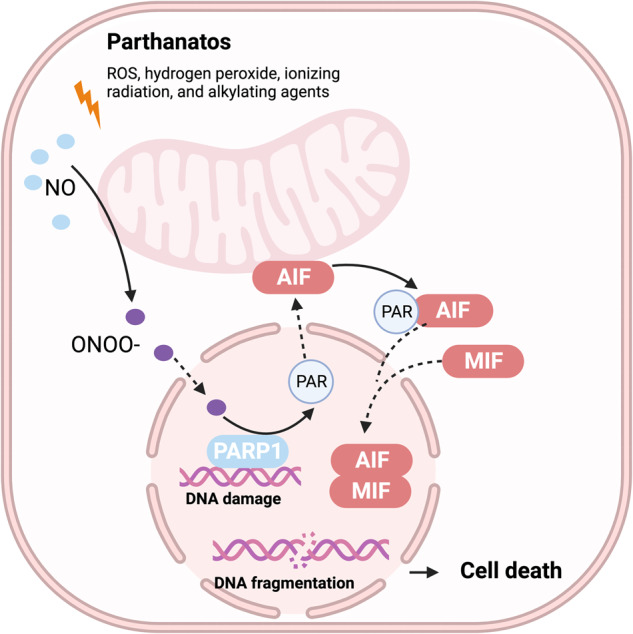
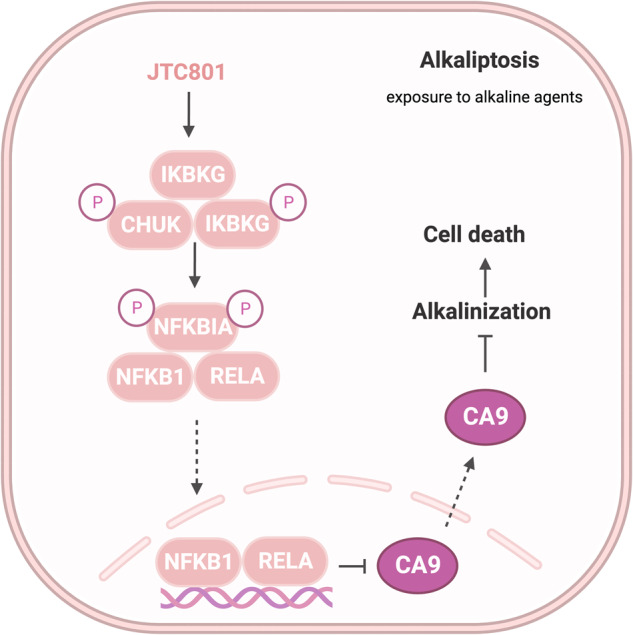
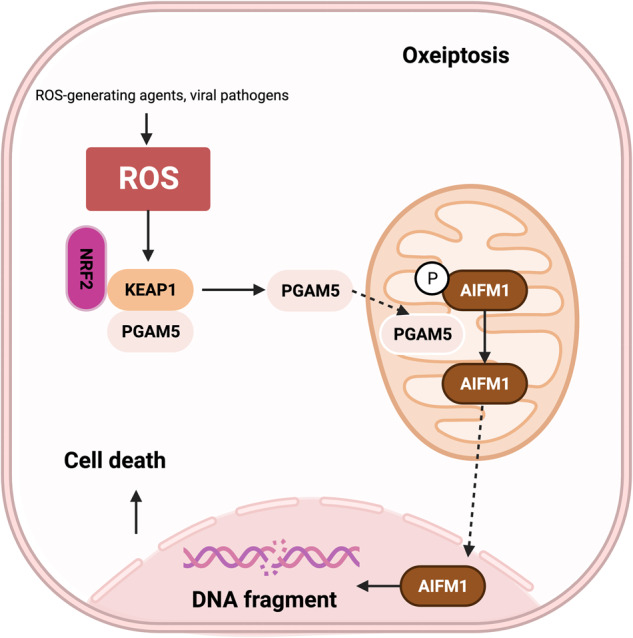
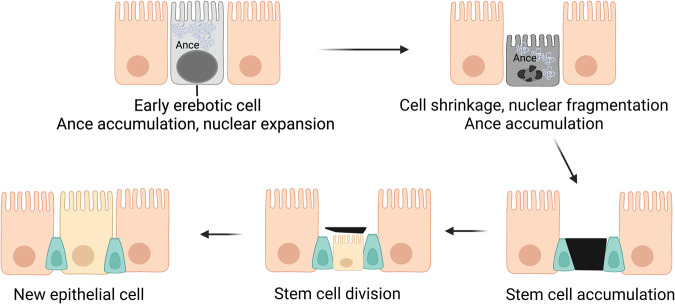
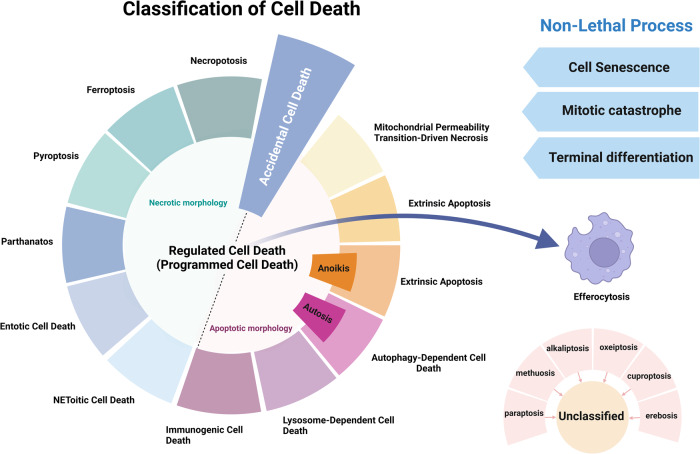
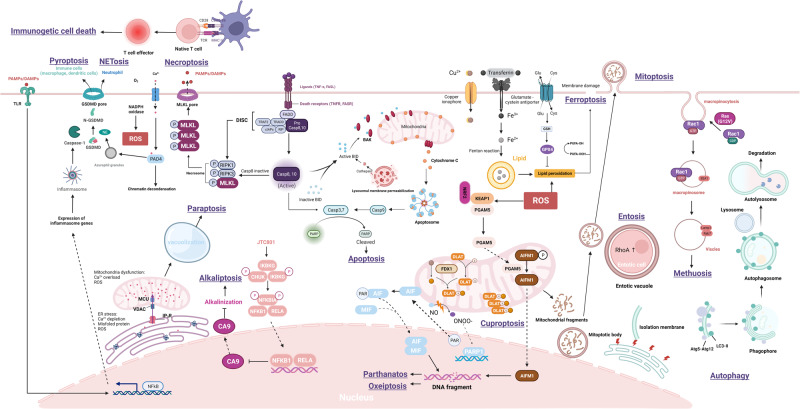
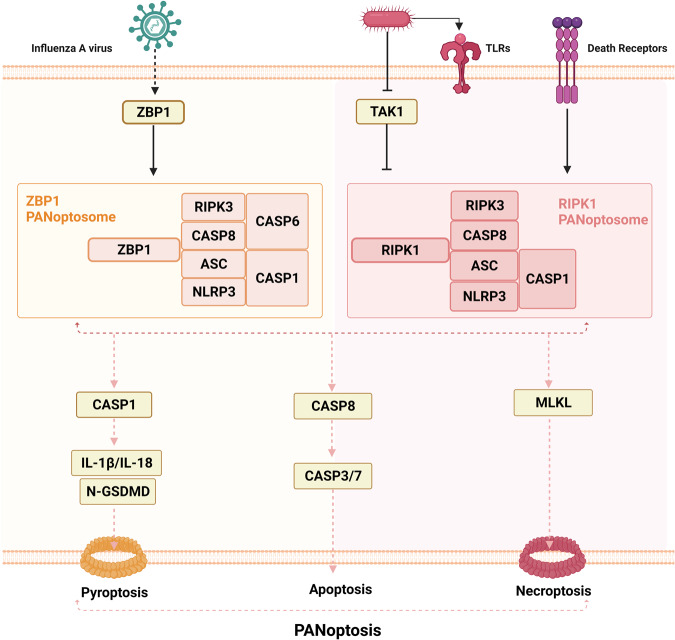
Death is the inevitable fate of all living organisms, whether at the individual or cellular level. For a long time, cell death was believed to be an undesirable but unavoidable final outcome of nonfunctioning cells, as inflammation was inevitably triggered in response to damage. However, experimental evidence accumulated over the past few decades has revealed different types of cell death that are genetically programmed to eliminate unnecessary or severely damaged cells that may damage surrounding tissues. Several types of cell death, including apoptosis, necrosis, autophagic cell death, and lysosomal cell death, which are classified as programmed cell death, and pyroptosis, necroptosis, and NETosis, which are classified as inflammatory cell death, have been described over the years. Recently, several novel forms of cell death, namely, mitoptosis, paraptosis, immunogenic cell death, entosis, methuosis, parthanatos, ferroptosis, autosis, alkaliptosis, oxeiptosis, cuproptosis, and erebosis, have been discovered and advanced our understanding of cell death and its complexity. In this review, we provide a historical overview of the discovery and characterization of different forms of cell death and highlight their diversity and complexity. We also briefly discuss the regulatory mechanisms underlying each type of cell death and the implications of cell death in various physiological and pathological contexts. This review provides a comprehensive understanding of different mechanisms of cell death that can be leveraged to develop novel therapeutic strategies for various diseases.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
