

Non-cell-autonomous cancer progression from chromosomal instability
- PMID: 37612508
- PMCID: PMC10468402
- DOI: 10.1038/s41586-023-06464-z
Non-cell-autonomous cancer progression from chromosomal instability
Abstract
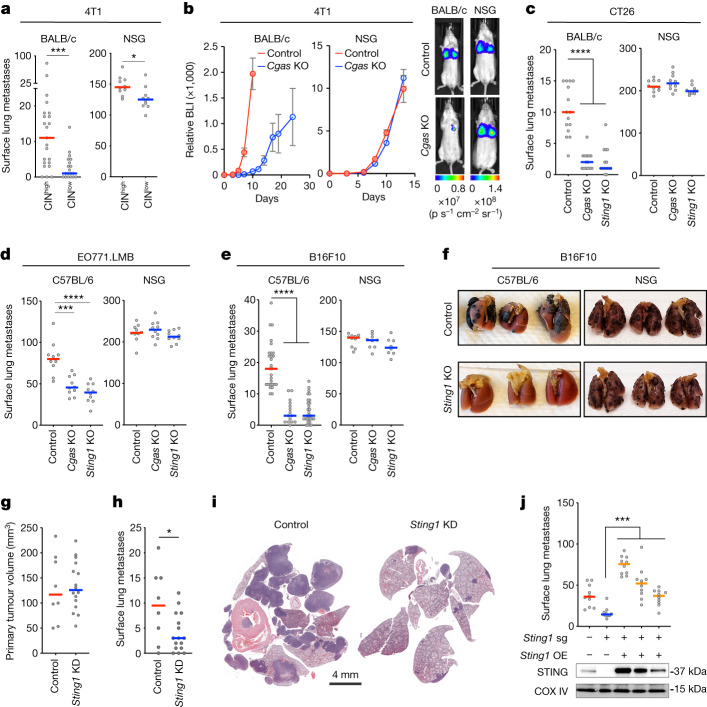
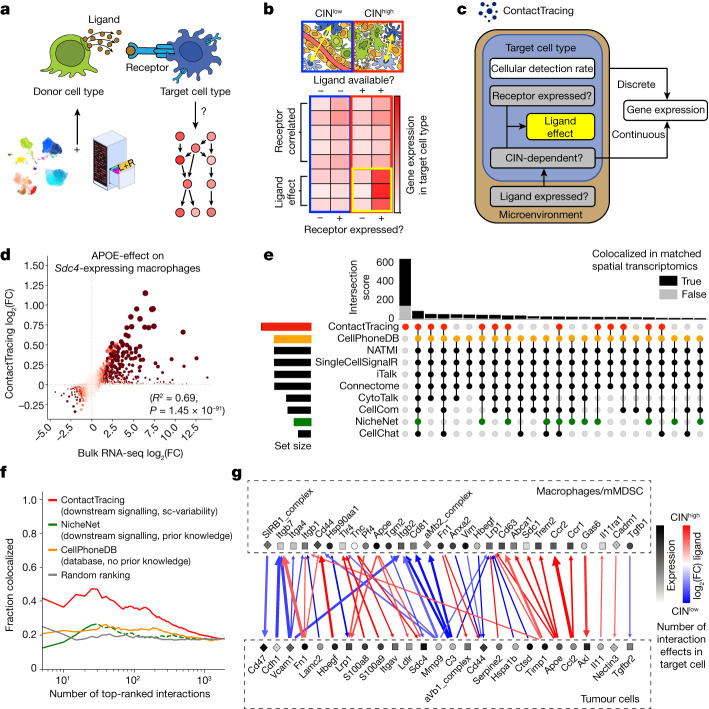
Chromosomal instability (CIN) is a driver of cancer metastasis1-4, yet the extent to which this effect depends on the immune system remains unknown. Using ContactTracing-a newly developed, validated and benchmarked tool to infer the nature and conditional dependence of cell-cell interactions from single-cell transcriptomic data-we show that CIN-induced chronic activation of the cGAS-STING pathway promotes downstream signal re-wiring in cancer cells, leading to a pro-metastatic tumour microenvironment. This re-wiring is manifested by type I interferon tachyphylaxis selectively downstream of STING and a corresponding increase in cancer cell-derived endoplasmic reticulum (ER) stress response. Reversal of CIN, depletion of cancer cell STING or inhibition of ER stress response signalling abrogates CIN-dependent effects on the tumour microenvironment and suppresses metastasis in immune competent, but not severely immune compromised, settings. Treatment with STING inhibitors reduces CIN-driven metastasis in melanoma, breast and colorectal cancers in a manner dependent on tumour cell-intrinsic STING. Finally, we show that CIN and pervasive cGAS activation in micronuclei are associated with ER stress signalling, immune suppression and metastasis in human triple-negative breast cancer, highlighting a viable strategy to identify and therapeutically intervene in tumours spurred by CIN-induced inflammation.
© 2023. The Author(s).
Conflict of interest statement
S.F.B. holds a patent related to some of the work described targeting CIN and the cGAS–STING pathway in advanced cancer. He owns equity in, receives compensation from, serves as a consultant for, and serves on the Scientific Advisory Board (SAB) and Board of Directors (BOD) of Volastra Therapeutics Inc., and serves on the SAB of Meliora Therapeutics. E.E.P. has served as a consultant for Boehringer Ingleheim. B.I. is a consultant for or received honoraria from Volastra Therapeutics, Johnson & Johnson/Janssen, Novartis, Eisai, AstraZeneca and Merck. H.W. is an advisor for AstraZeneca and received honoraria from Roche. J.S.R.-F. consults for Goldman Sachs, Bain Capital, Repare Therapeutics, Personalis, Saga Diagnostics and Paige.AI; serves on the SAB of Repare Therapeutics, VolitionRx, Paige.AI, AstraZeneca, MSD, Personalis and Daiichi Sankyo; and serves on the BOD of Grupo Oncoclínicas. S.B., J.J.H. and B.T. are employees of and own equity in Volastra Therapeutics Inc. C.G. consults for Cellino Biotech and Gardian Bio. The remaining authors declare no competing interests.
Figures















Comment in
-
Chromosomal instability drives immunosuppression and metastatic dissemination.Cell Res. 2024 Jan;34(1):7-8. doi: 10.1038/s41422-023-00876-2. Cell Res. 2024. PMID: 37739994 Free PMC article. No abstract available.
-
Chromosomal instability (CIN) triggers immune evasion and metastatic potential in cancer through rewired STING signalling.Mol Biomed. 2024 Jan 23;5(1):4. doi: 10.1186/s43556-023-00166-8. Mol Biomed. 2024. PMID: 38253764 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R37 CA258829/CA/NCI NIH HHS/United States
- R01 CA280414/CA/NCI NIH HHS/United States
- R21 CA263381/CA/NCI NIH HHS/United States
- R01 CA280572/CA/NCI NIH HHS/United States
- R21 CA266660/CA/NCI NIH HHS/United States
- DP5 OD026395/OD/NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- P50 CA247749/CA/NCI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 CA266446/CA/NCI NIH HHS/United States
- T32 GM132083/GM/NIGMS NIH HHS/United States
- R01 CA256188/CA/NCI NIH HHS/United States
- U01 CA210152/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
