

Assembly of 43 human Y chromosomes reveals extensive complexity and variation
- PMID: 37612510
- PMCID: PMC10726138
- DOI: 10.1038/s41586-023-06425-6
Assembly of 43 human Y chromosomes reveals extensive complexity and variation
Abstract
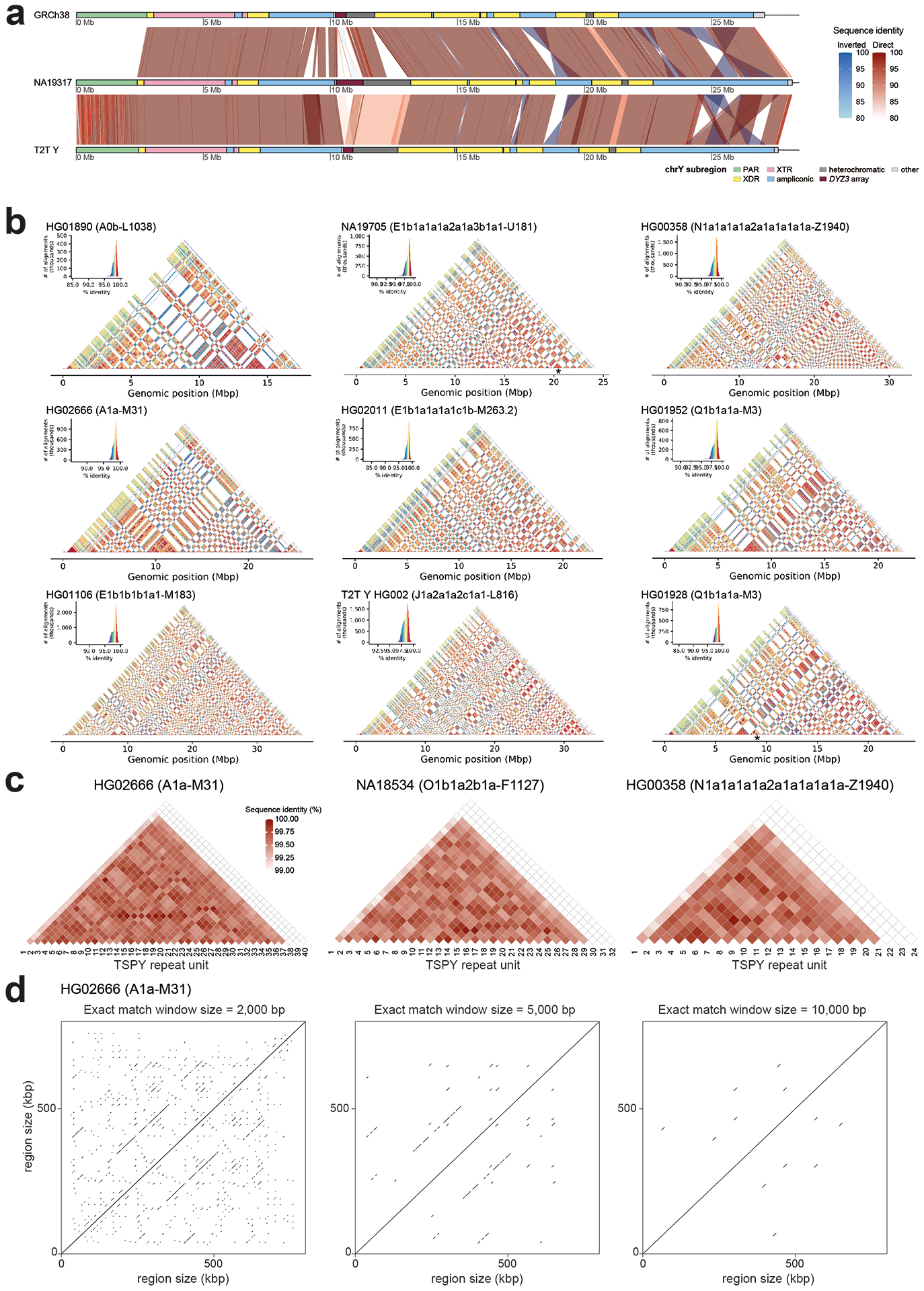
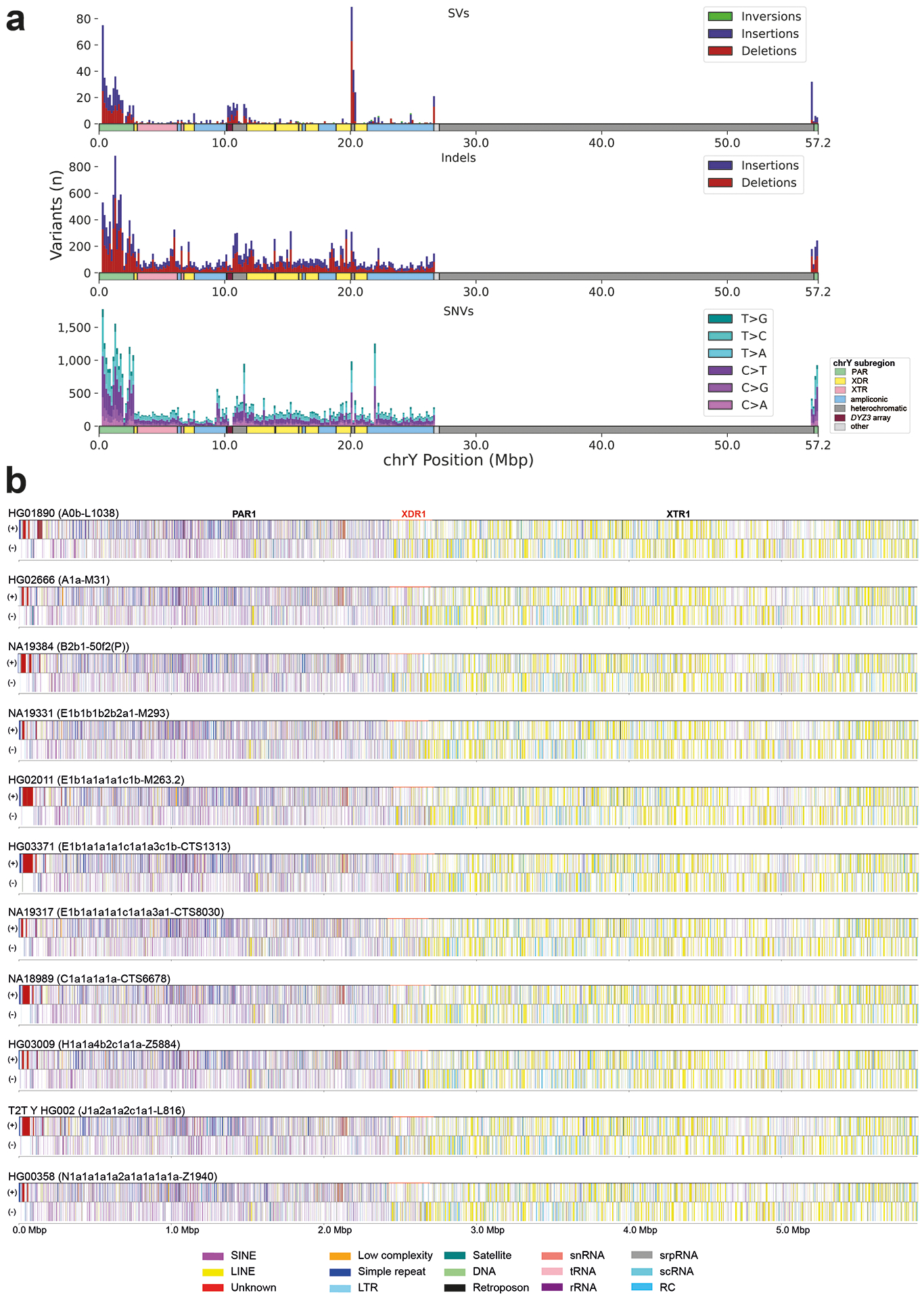
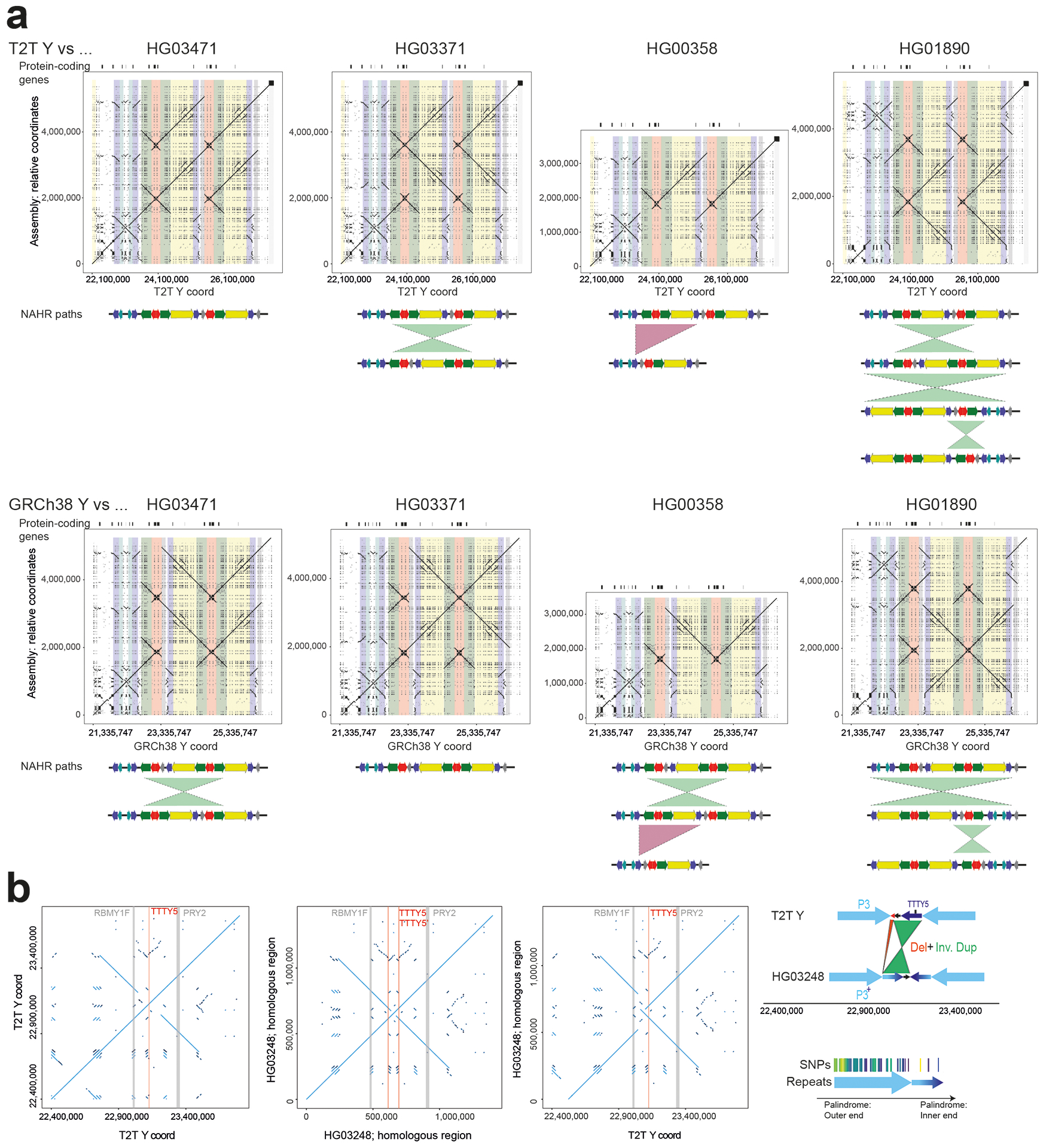
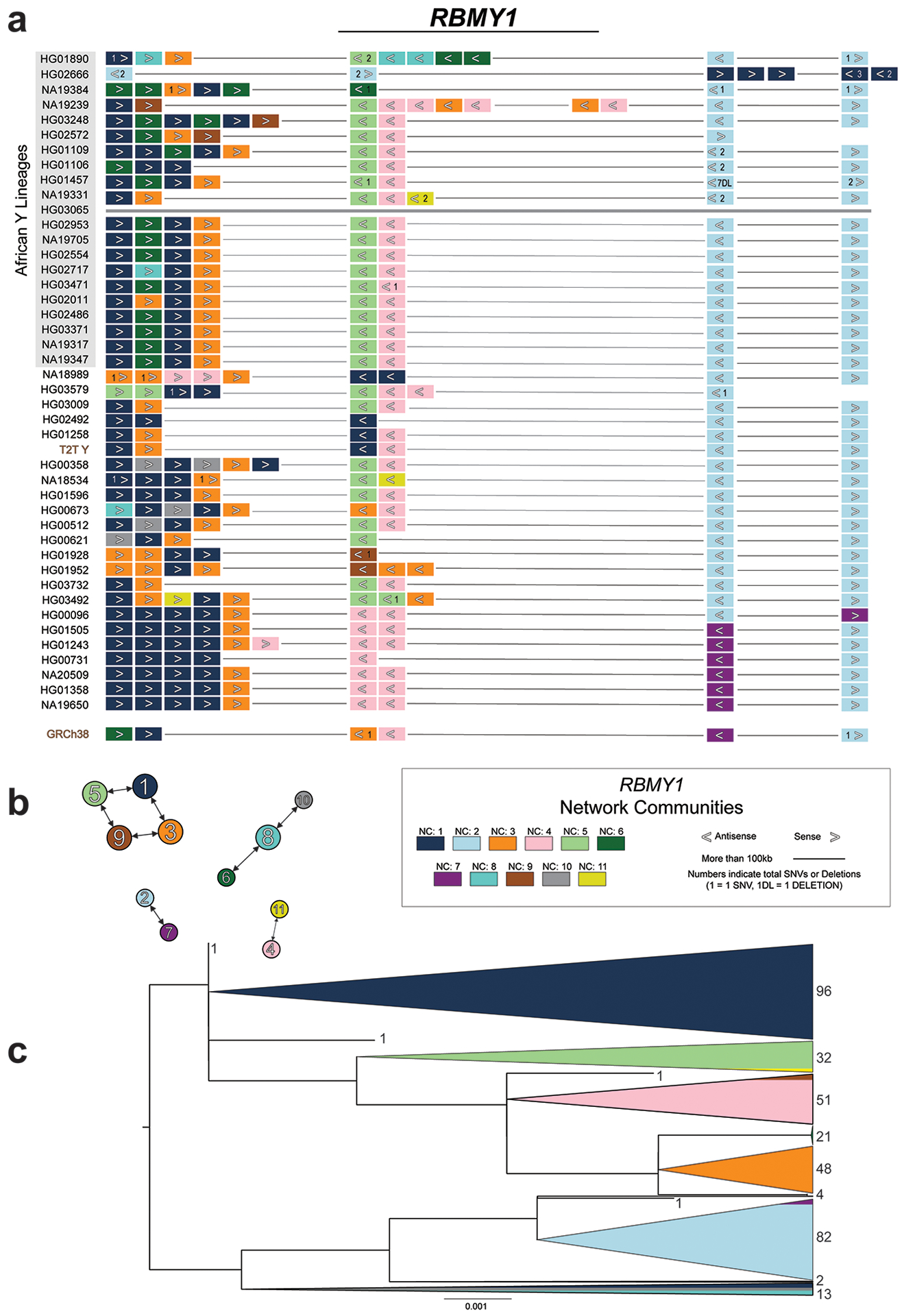
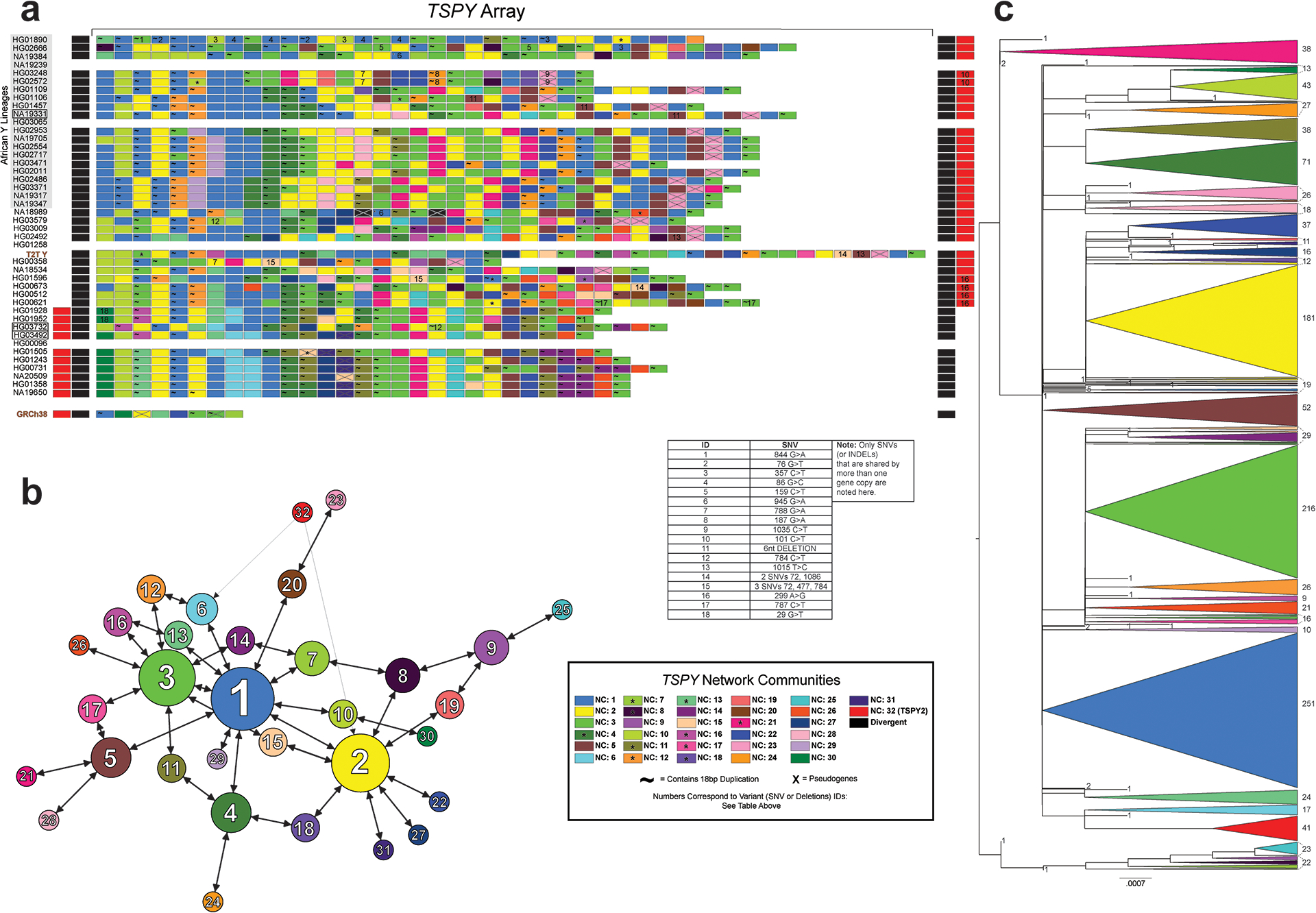
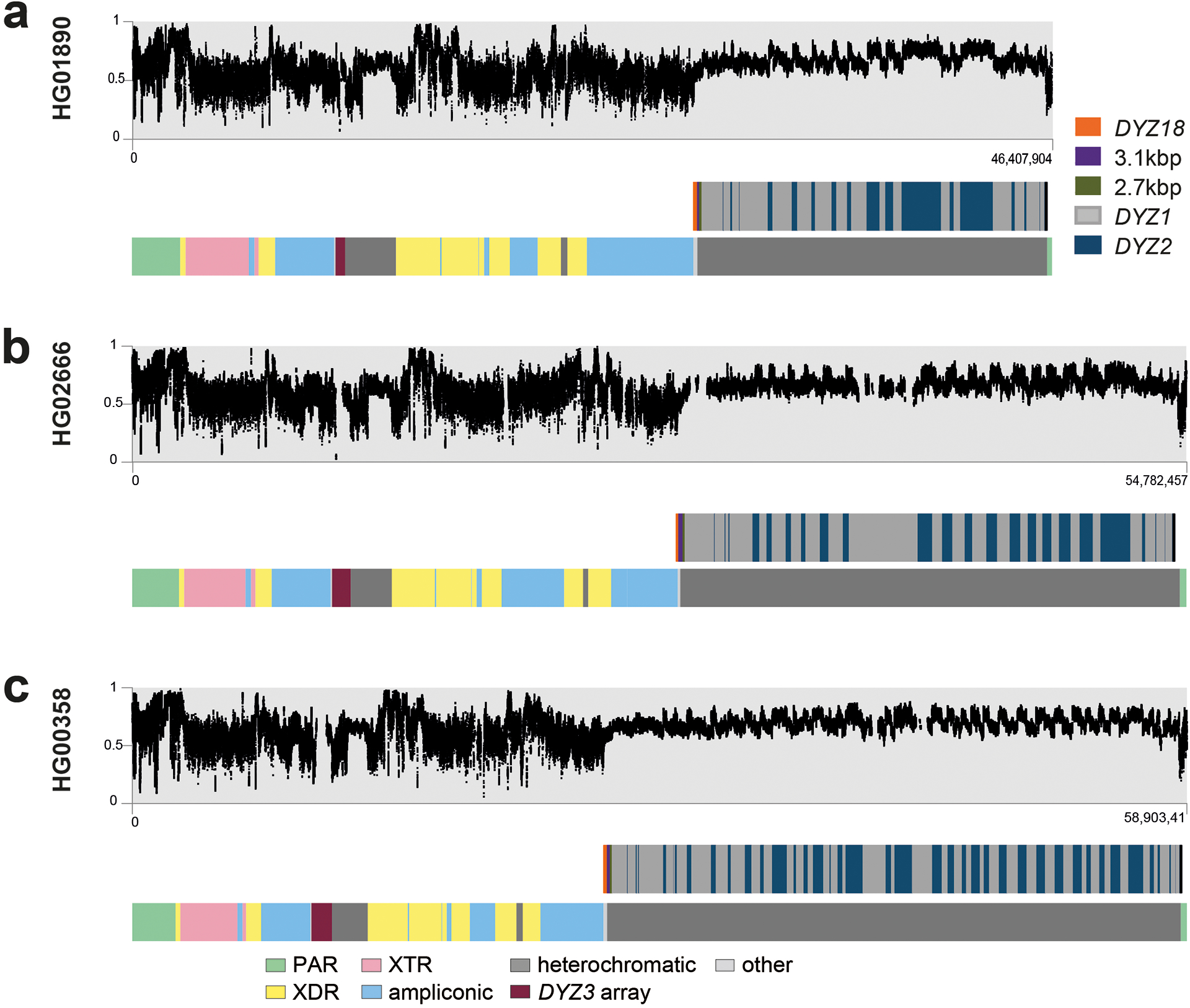
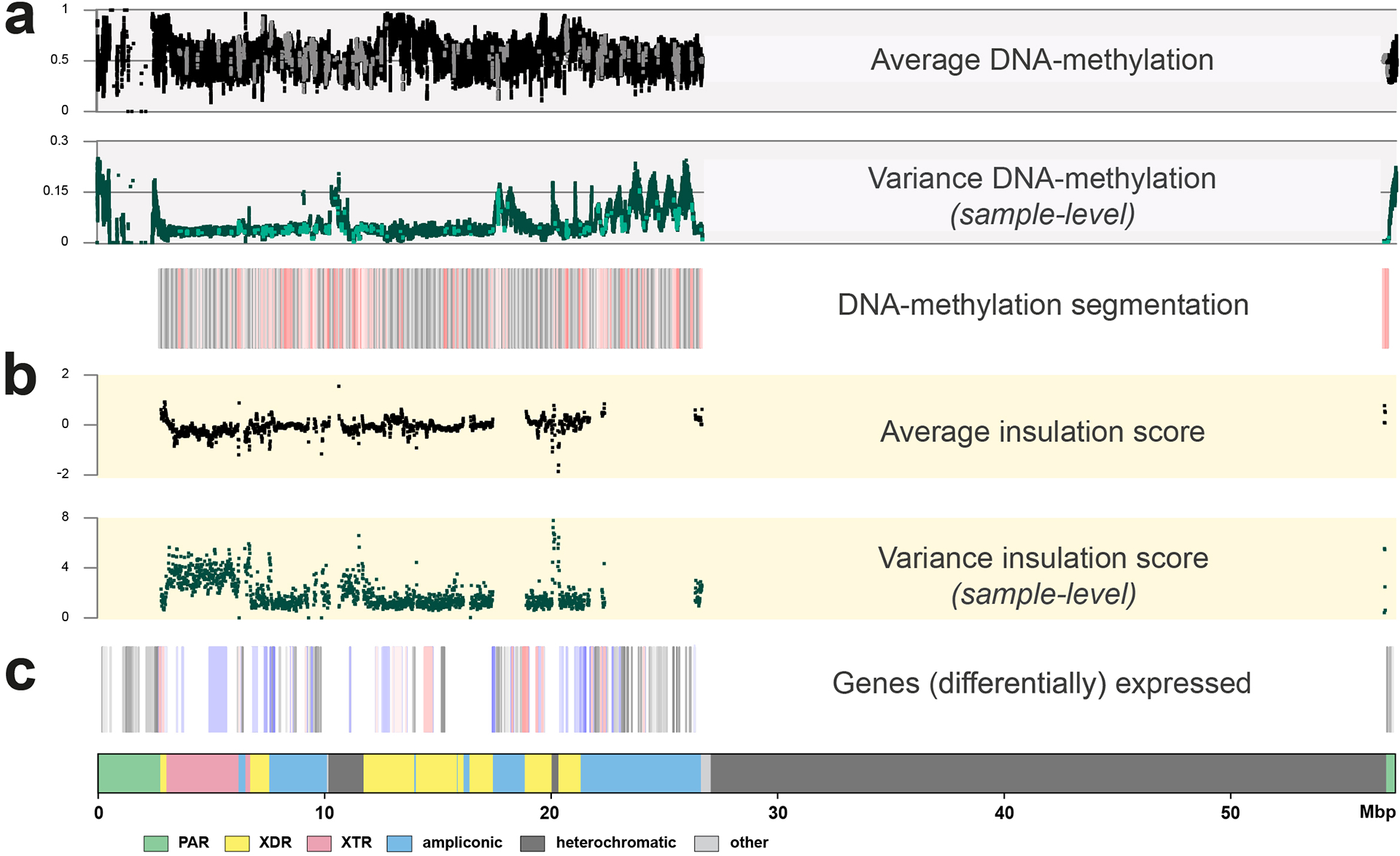
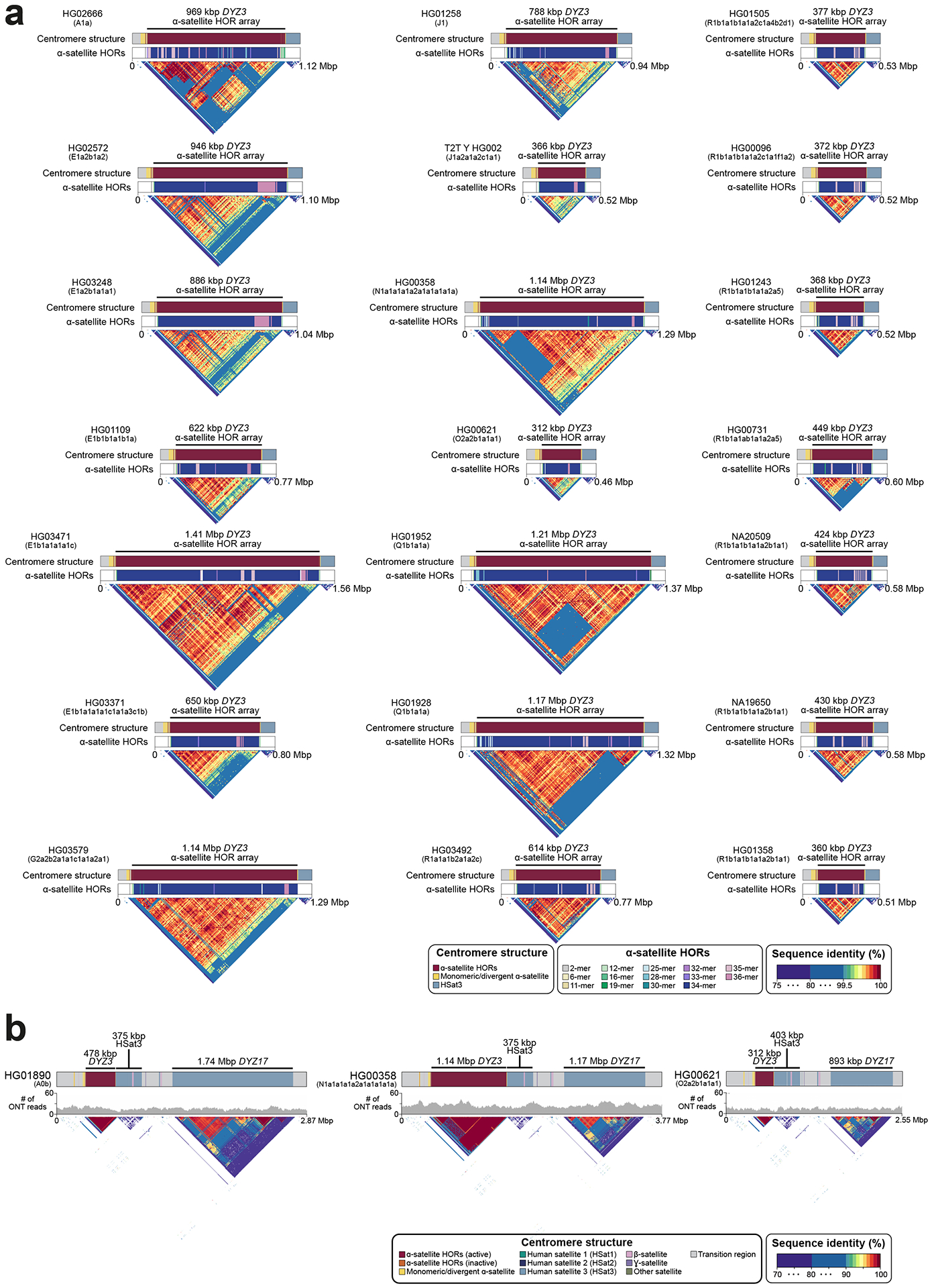
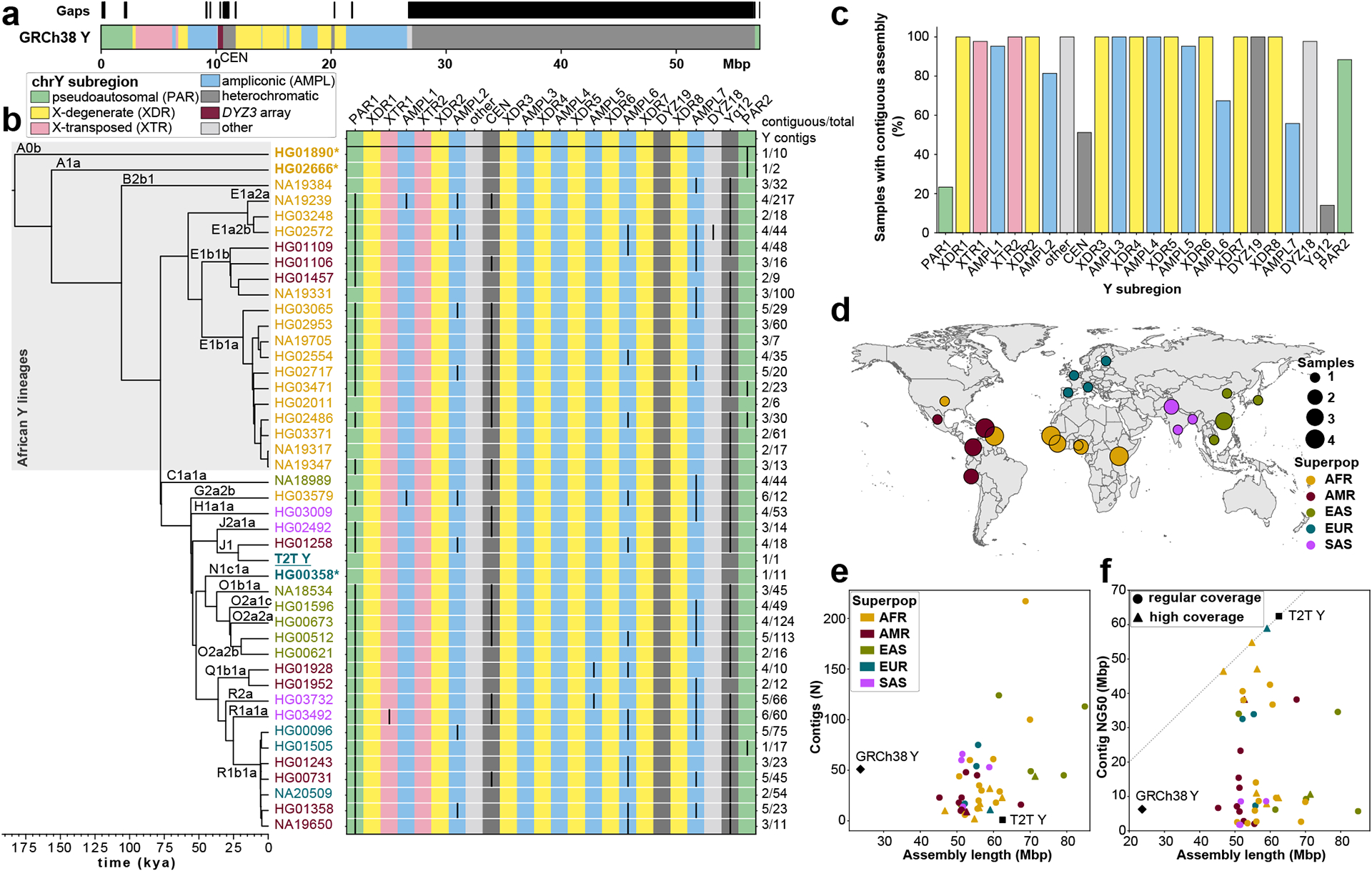
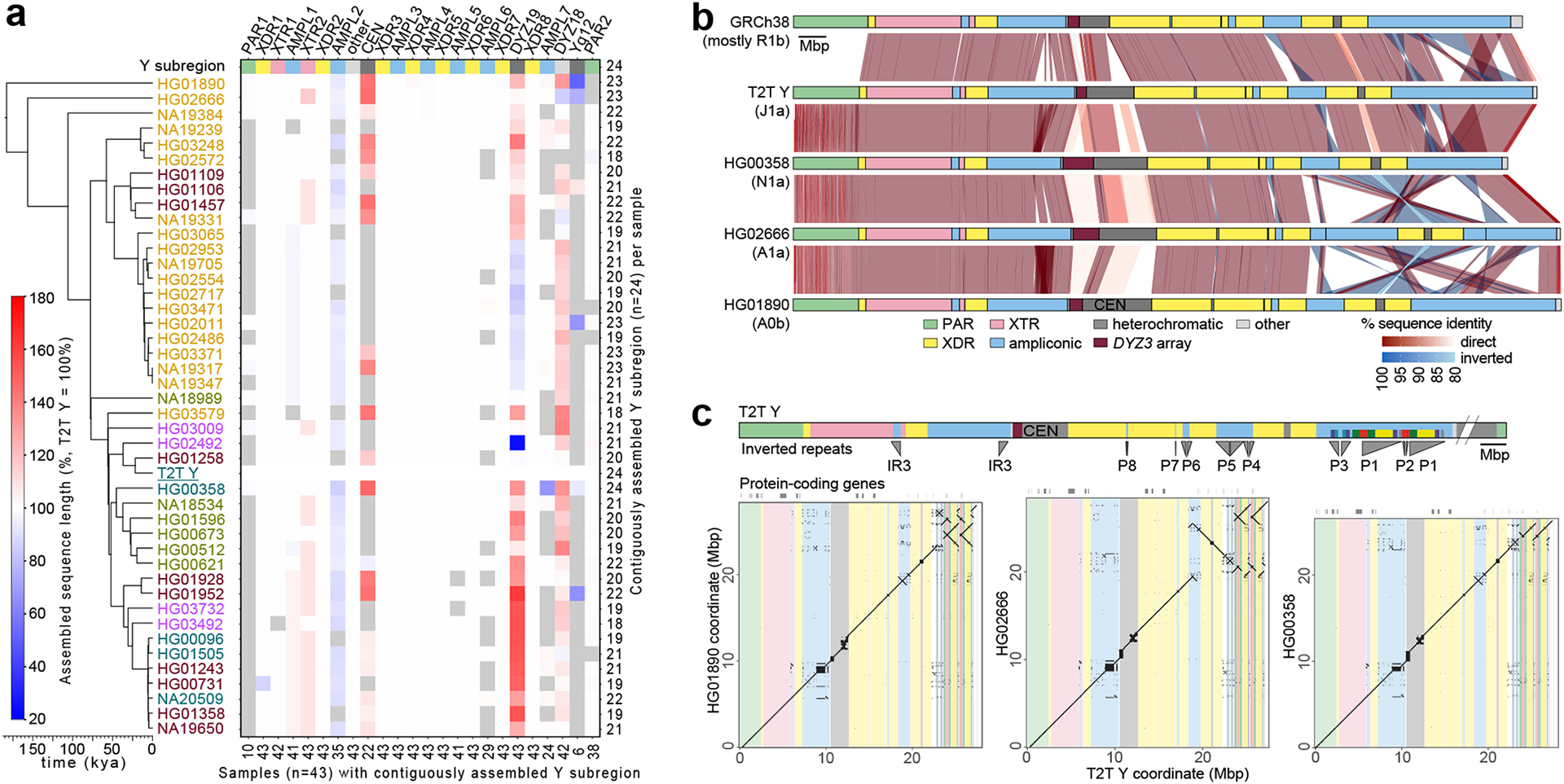
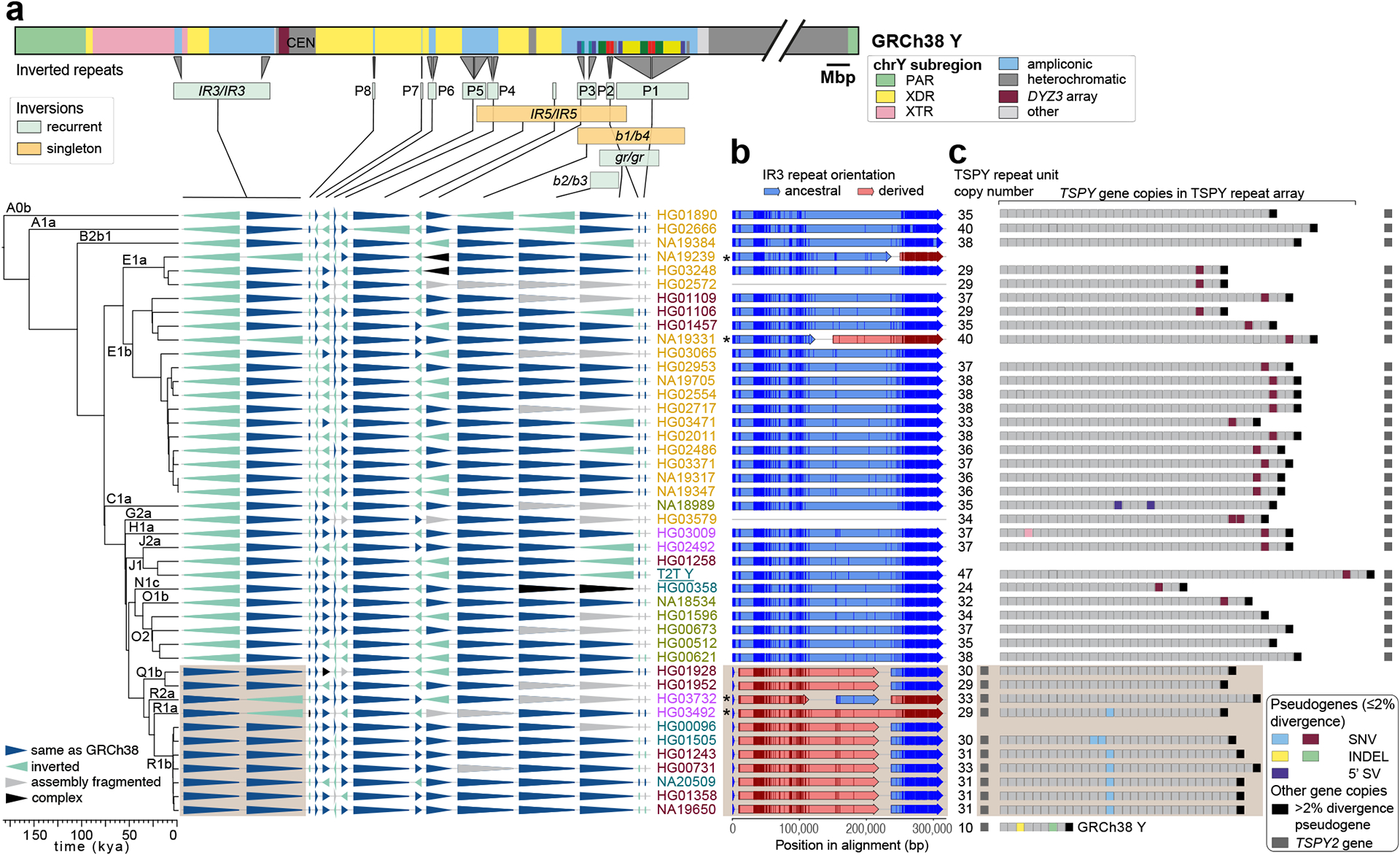
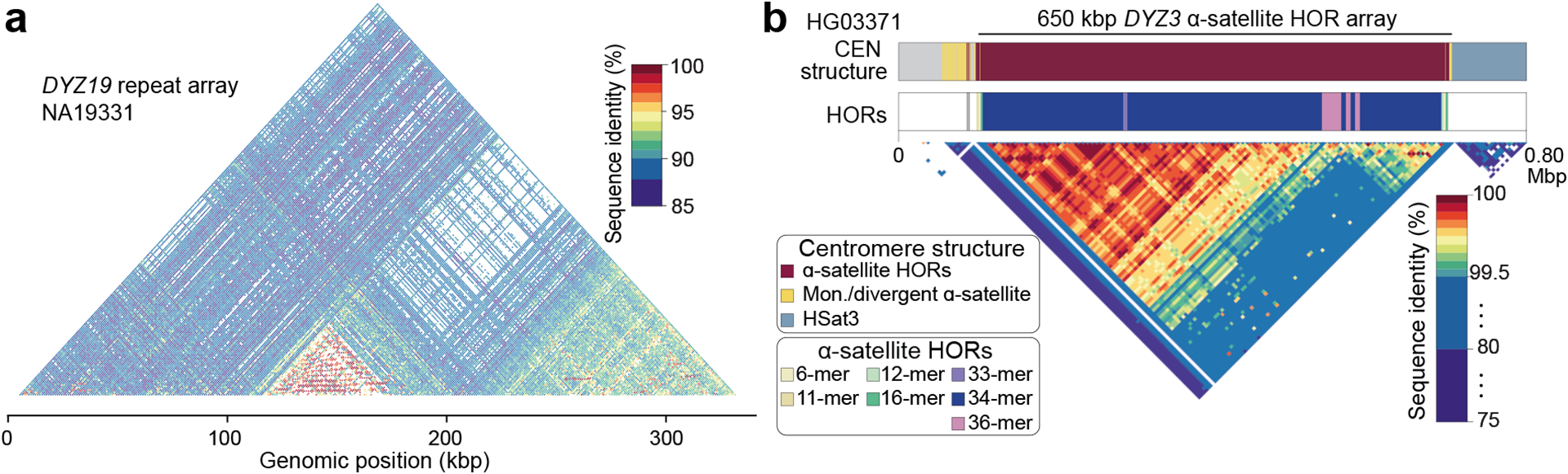
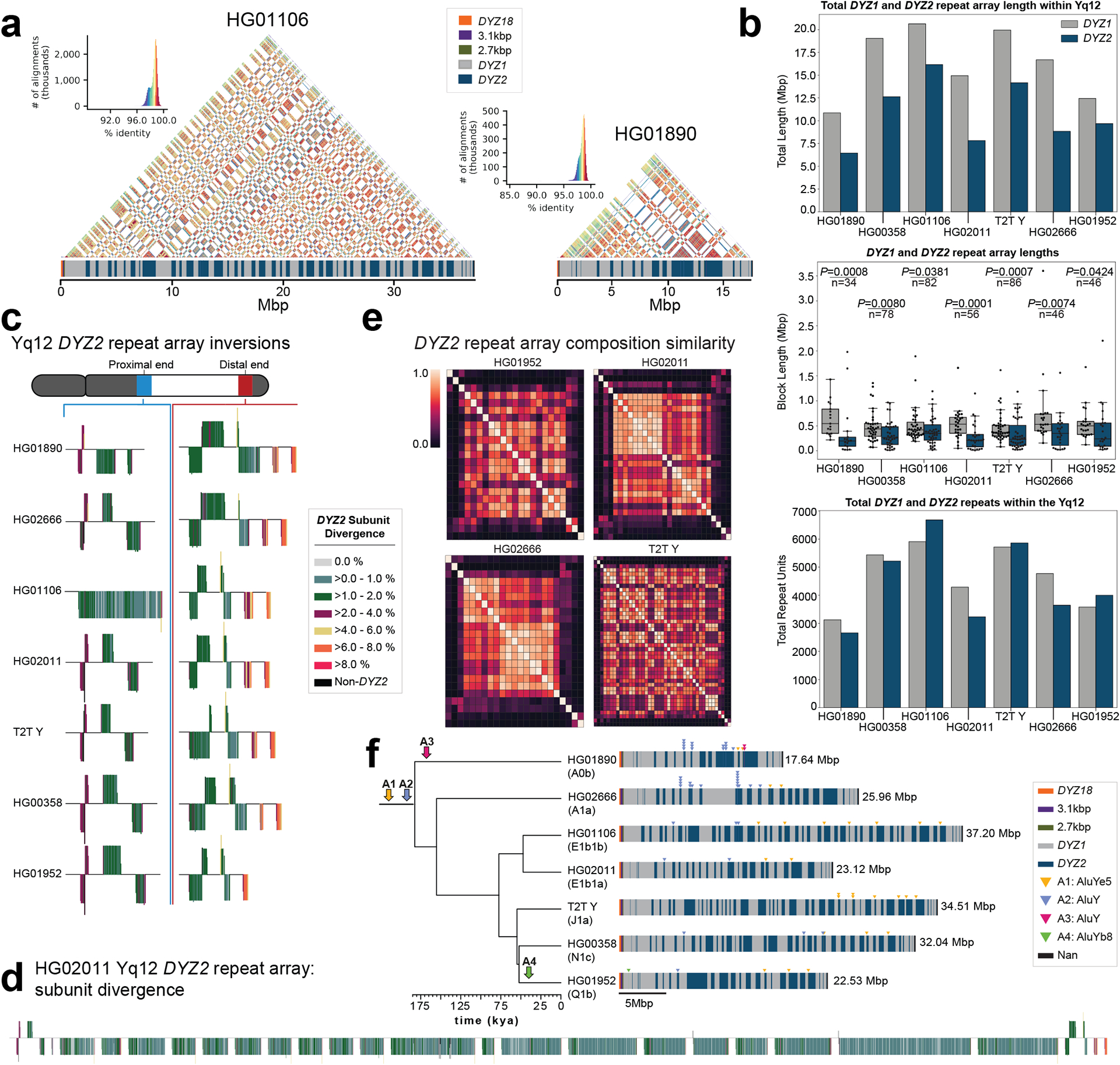
The prevalence of highly repetitive sequences within the human Y chromosome has prevented its complete assembly to date1 and led to its systematic omission from genomic analyses. Here we present de novo assemblies of 43 Y chromosomes spanning 182,900 years of human evolution and report considerable diversity in size and structure. Half of the male-specific euchromatic region is subject to large inversions with a greater than twofold higher recurrence rate compared with all other chromosomes2. Ampliconic sequences associated with these inversions show differing mutation rates that are sequence context dependent, and some ampliconic genes exhibit evidence for concerted evolution with the acquisition and purging of lineage-specific pseudogenes. The largest heterochromatic region in the human genome, Yq12, is composed of alternating repeat arrays that show extensive variation in the number, size and distribution, but retain a 1:1 copy-number ratio. Finally, our data suggest that the boundary between the recombining pseudoautosomal region 1 and the non-recombining portions of the X and Y chromosomes lies 500 kb away from the currently established1 boundary. The availability of fully sequence-resolved Y chromosomes from multiple individuals provides a unique opportunity for identifying new associations of traits with specific Y-chromosomal variants and garnering insights into the evolution and function of complex regions of the human genome.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
E.E.E. is a scientific advisory board member of Variant Bio, Inc. C. Lee is a scientific advisory board member of Nabsys and Genome Insight. The following authors have previously disclosed a patent application (no. EP19169090) relevant to Strand-seq: J.O.K., T.M. and D.P. The other authors declare no competing interests.
Figures


Comment in
-
Unlocking the mystery of the human Y chromosome.Nat Rev Urol. 2024 Feb;21(2):65-66. doi: 10.1038/s41585-023-00826-y. Nat Rev Urol. 2024. PMID: 37803196 No abstract available.
References
-
- Skaletsky H et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
