

The complete sequence of a human Y chromosome
- PMID: 37612512
- PMCID: PMC10752217
- DOI: 10.1038/s41586-023-06457-y
The complete sequence of a human Y chromosome
Abstract
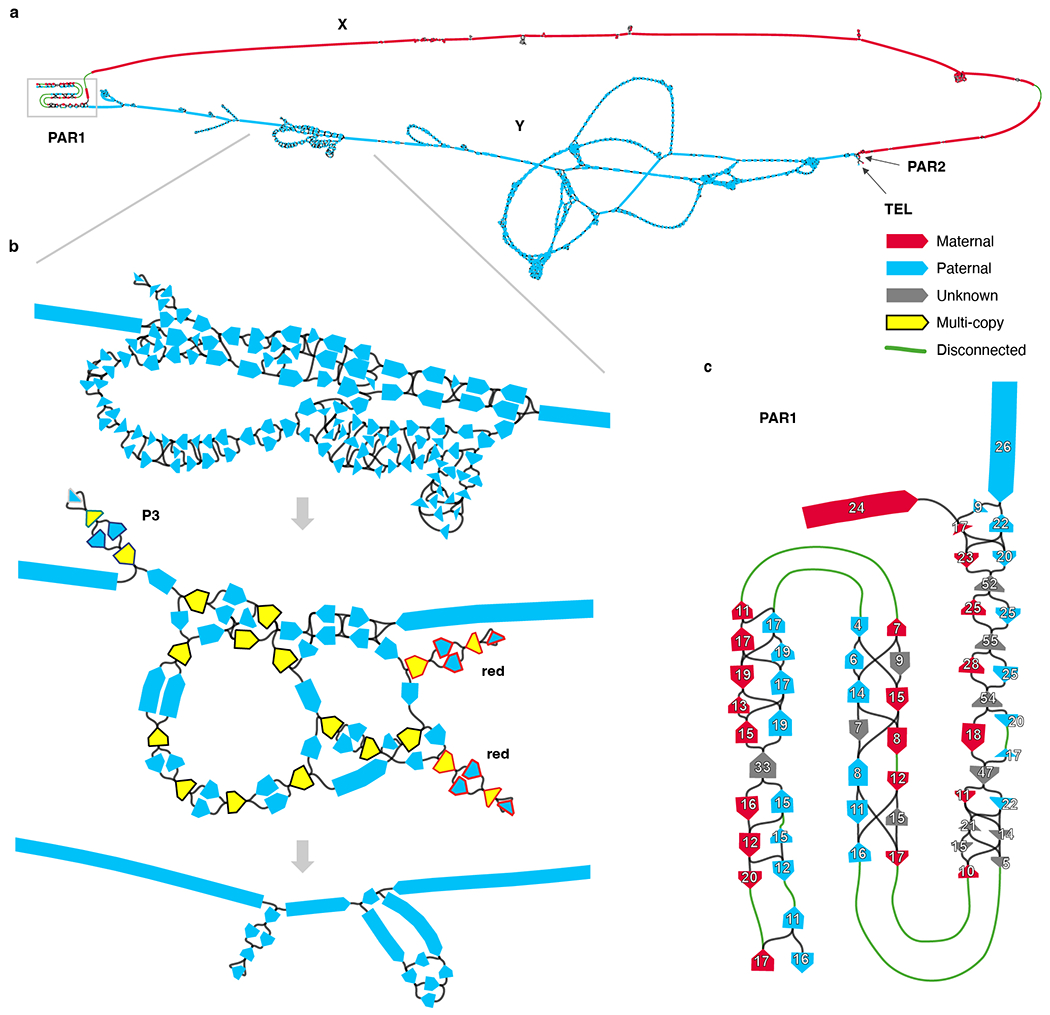
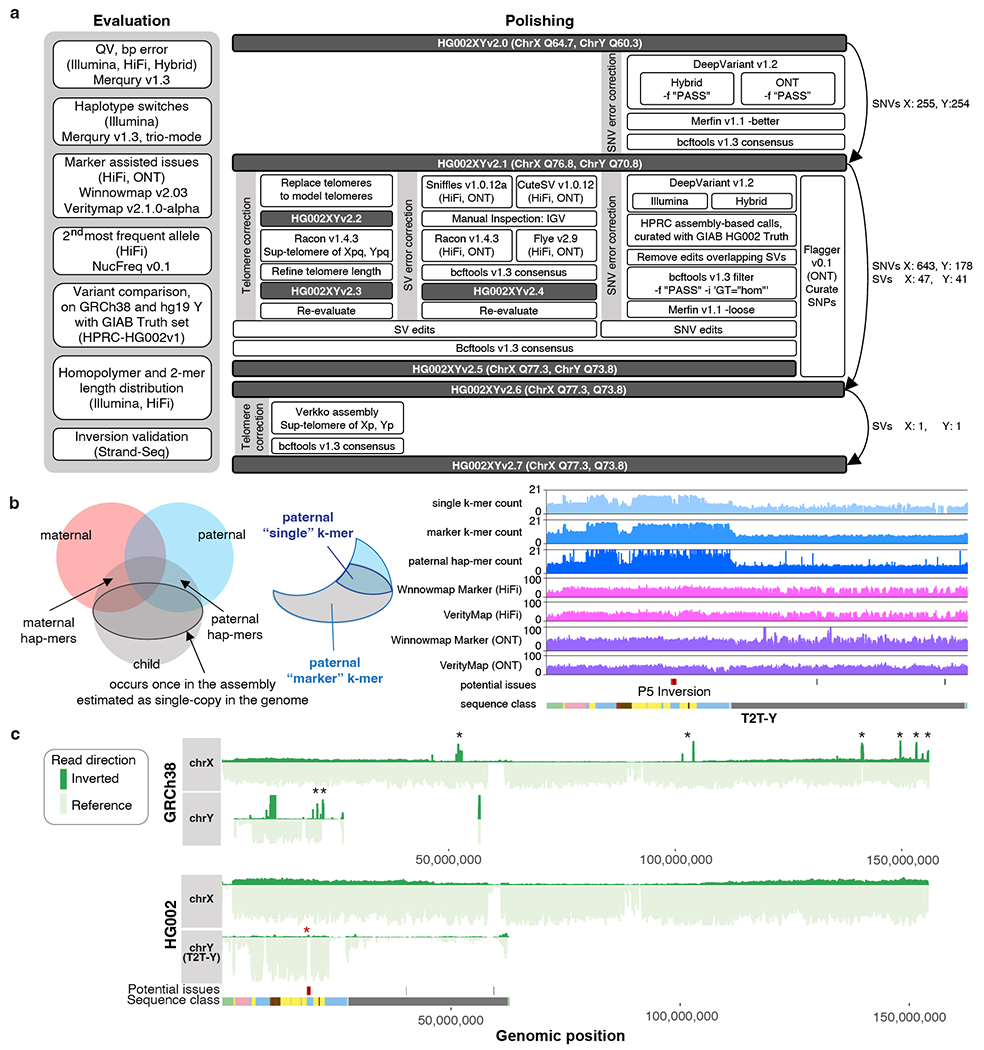
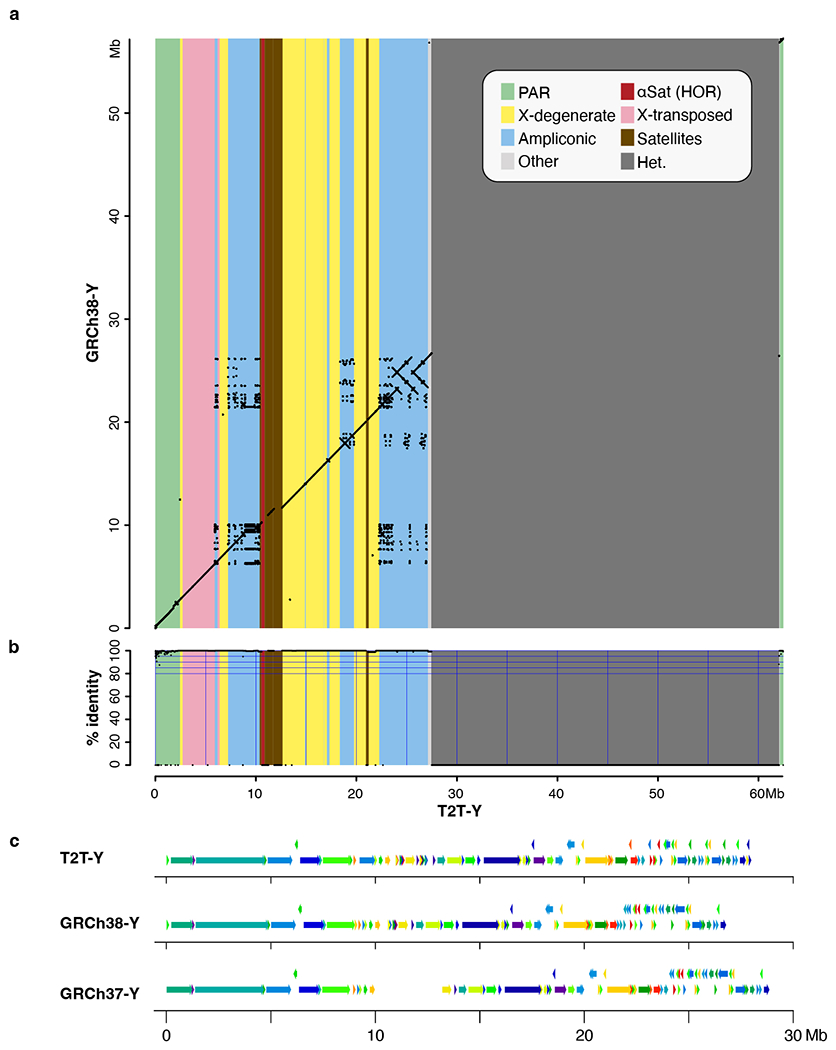
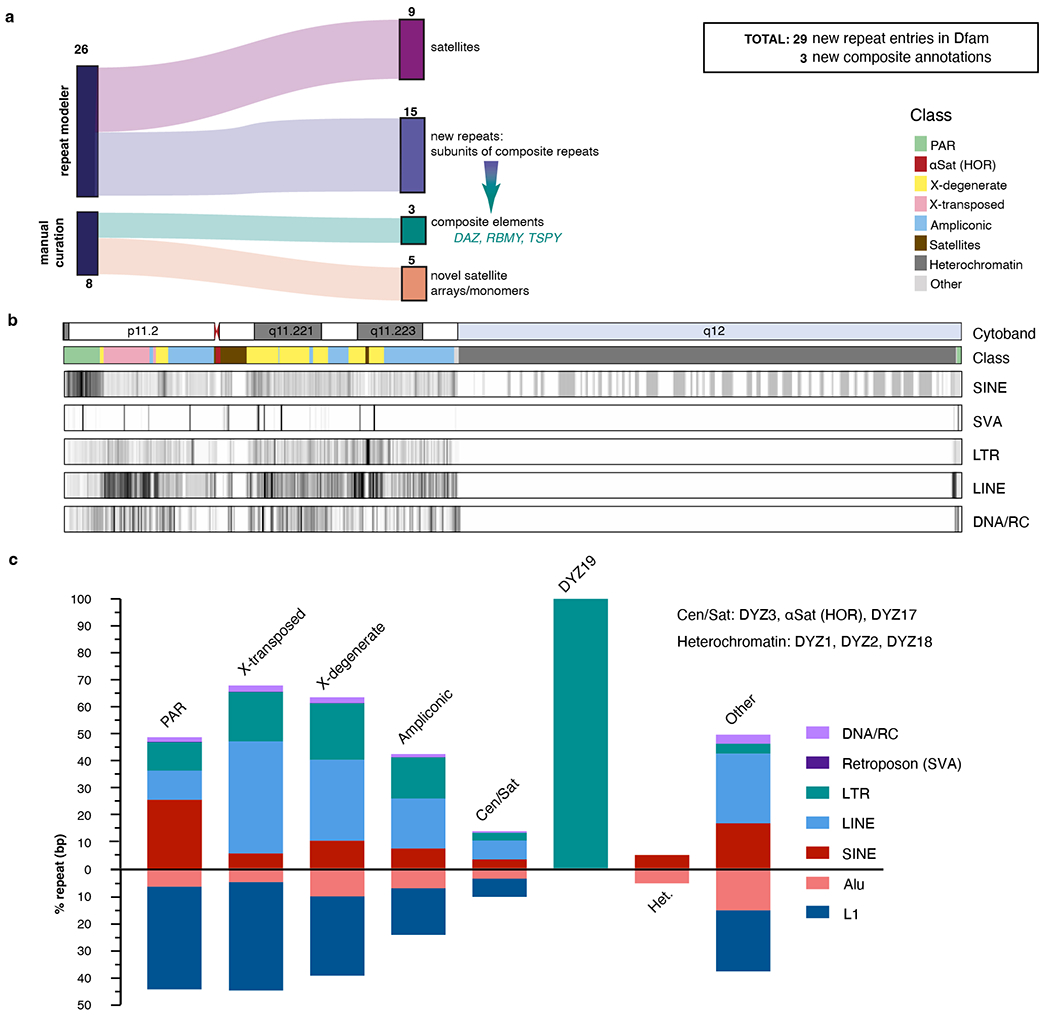
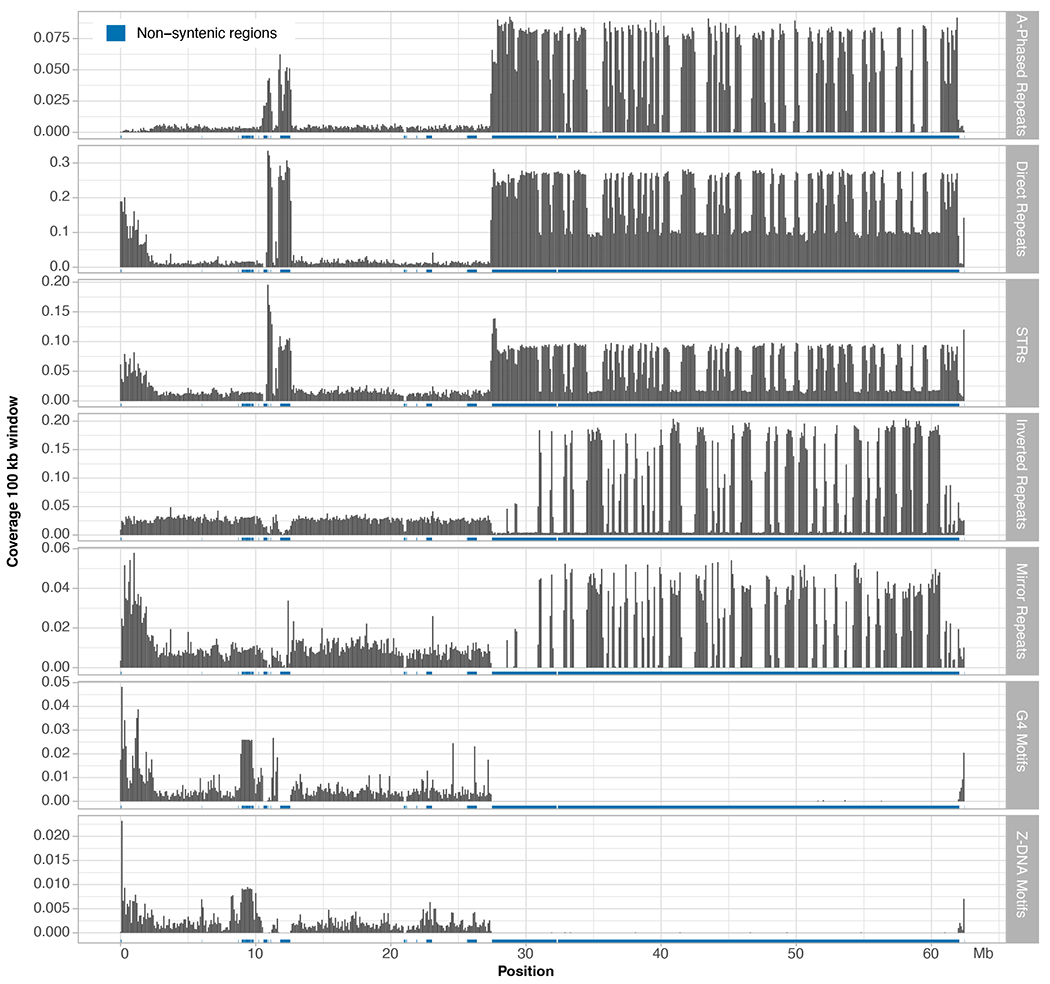
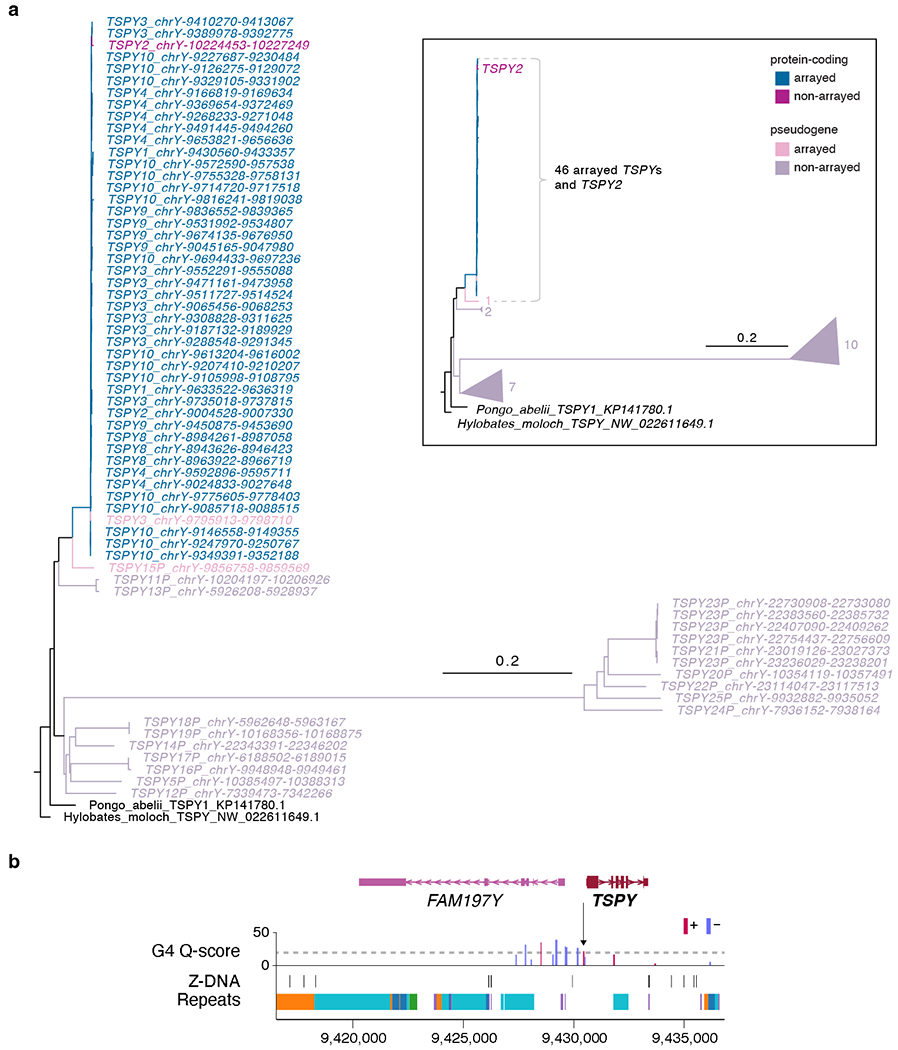
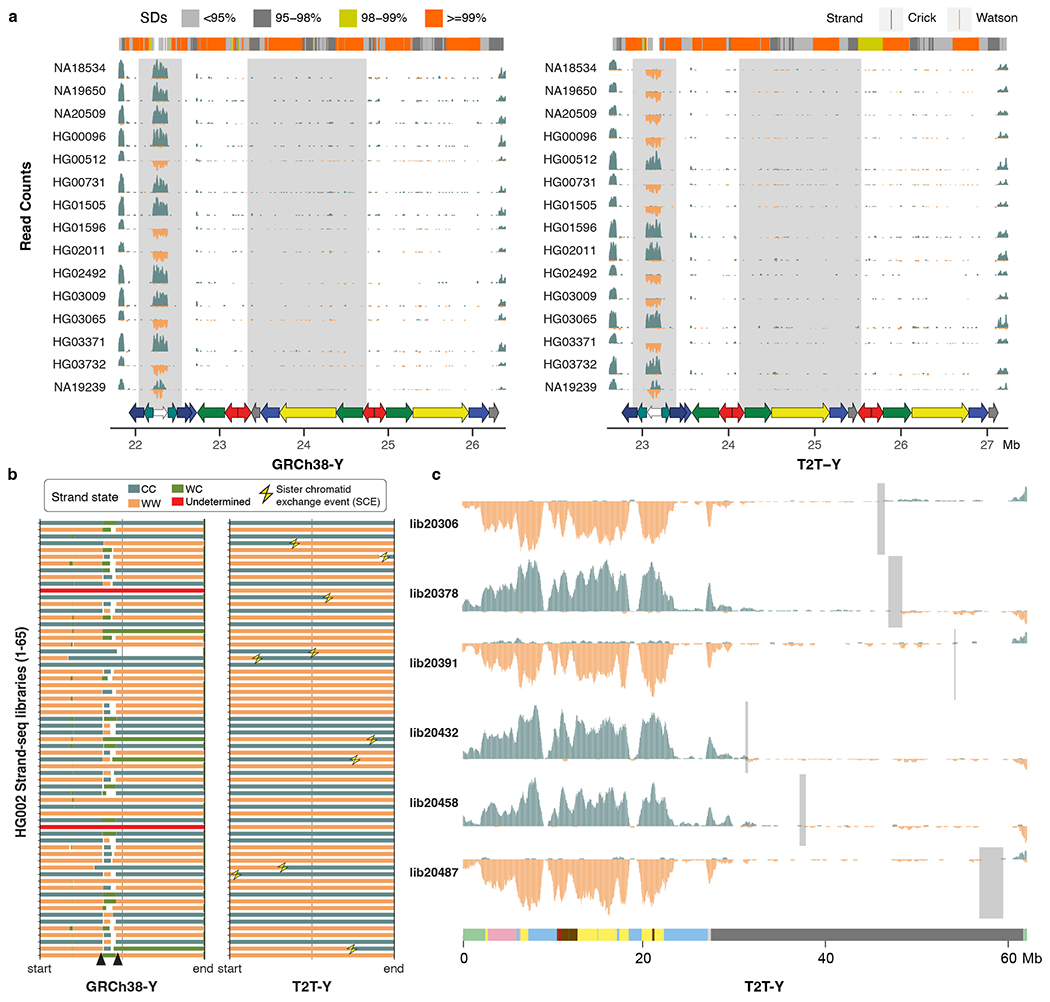
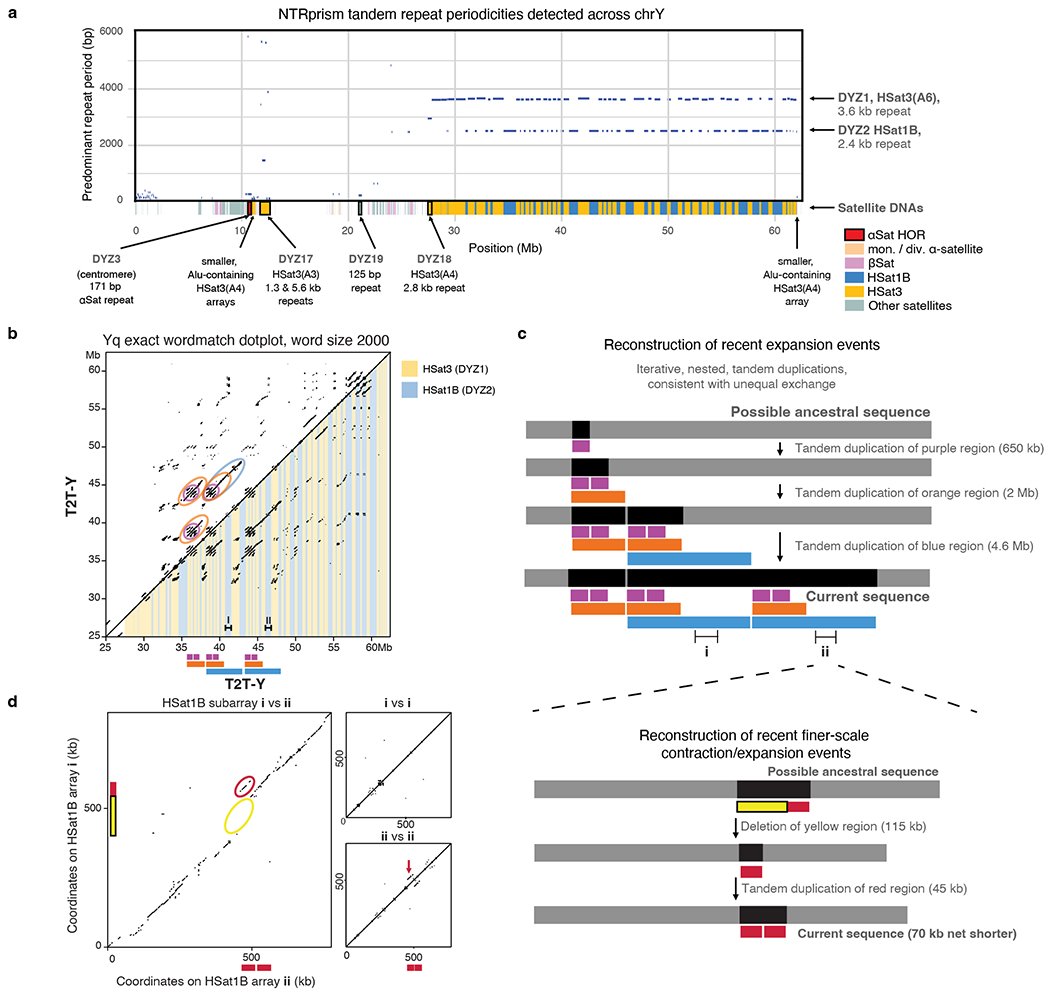
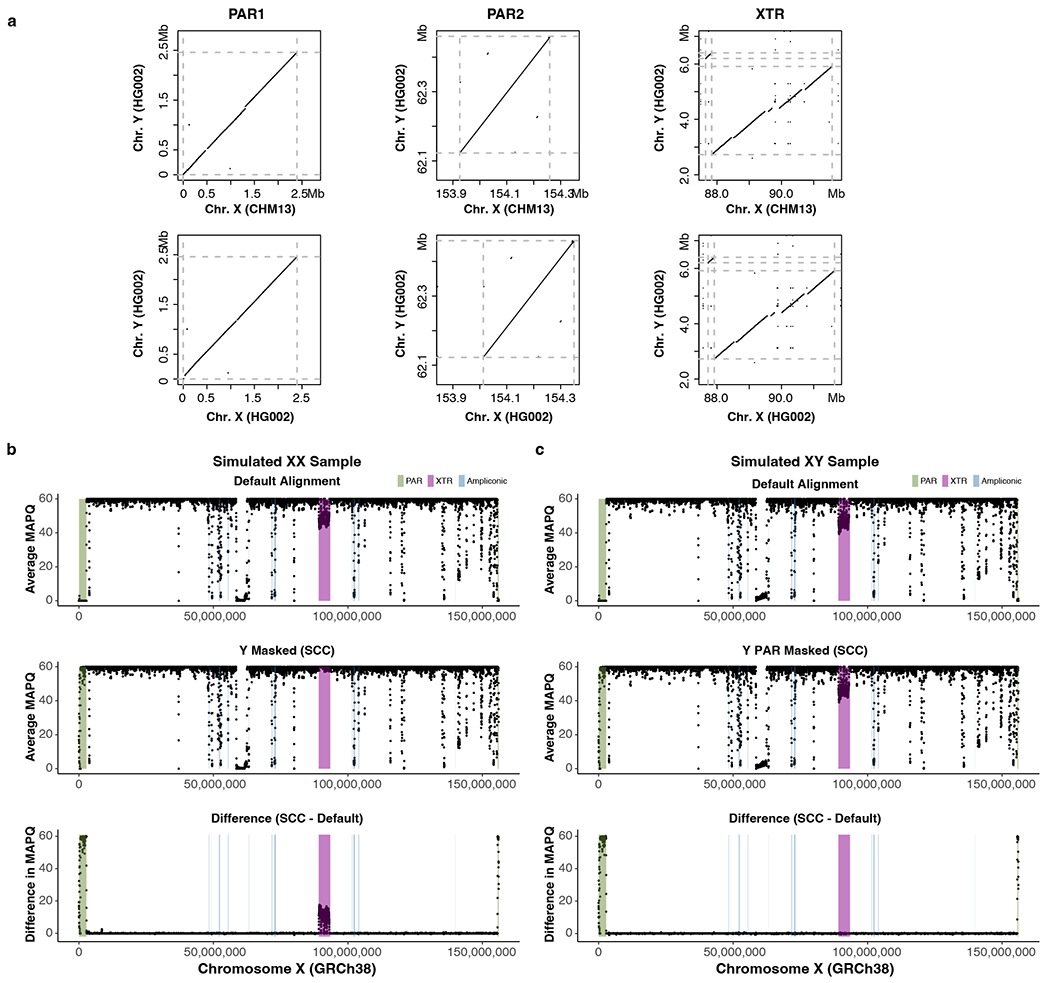
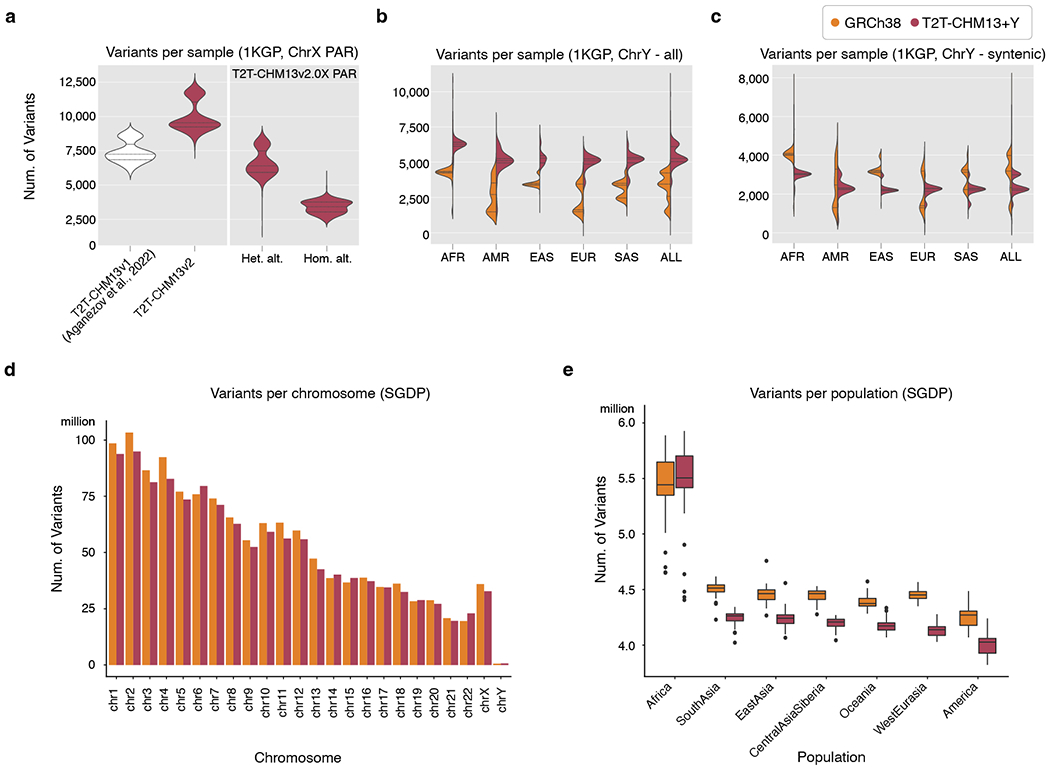
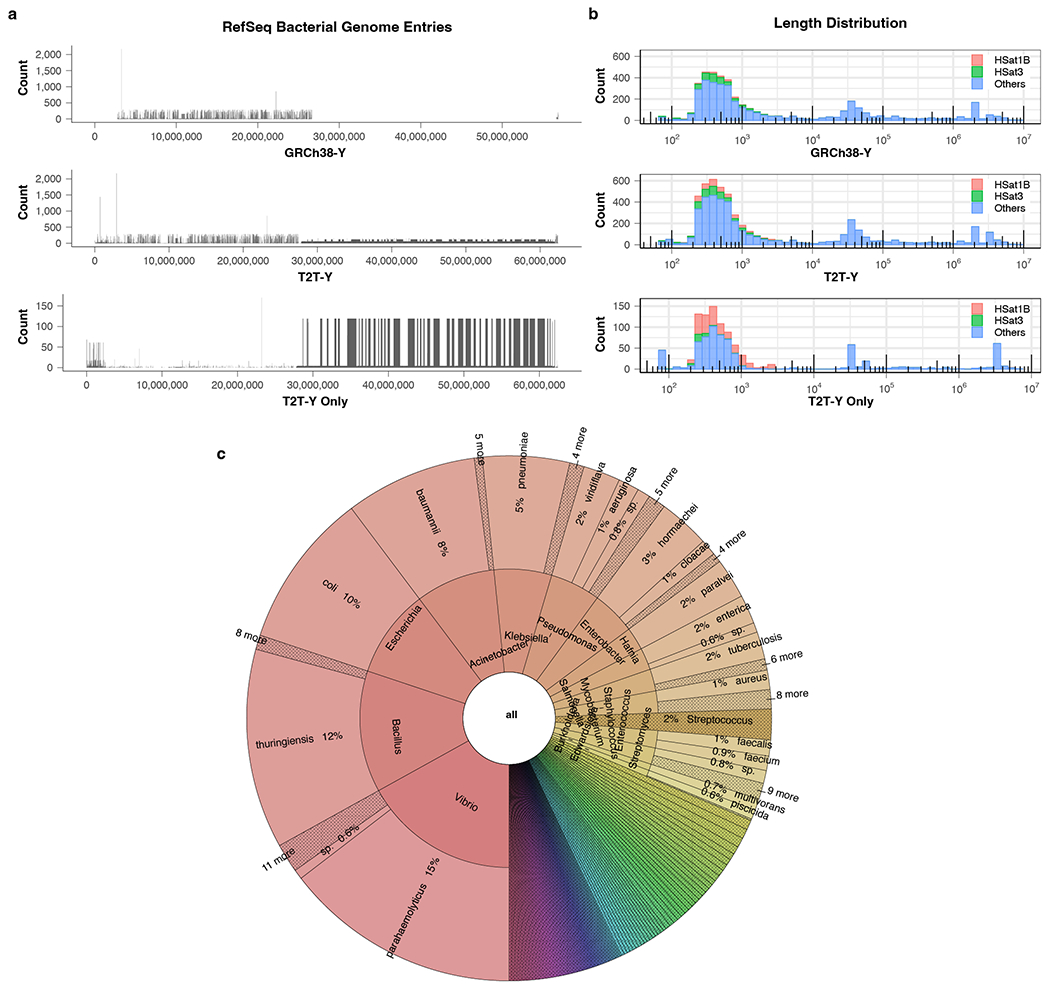
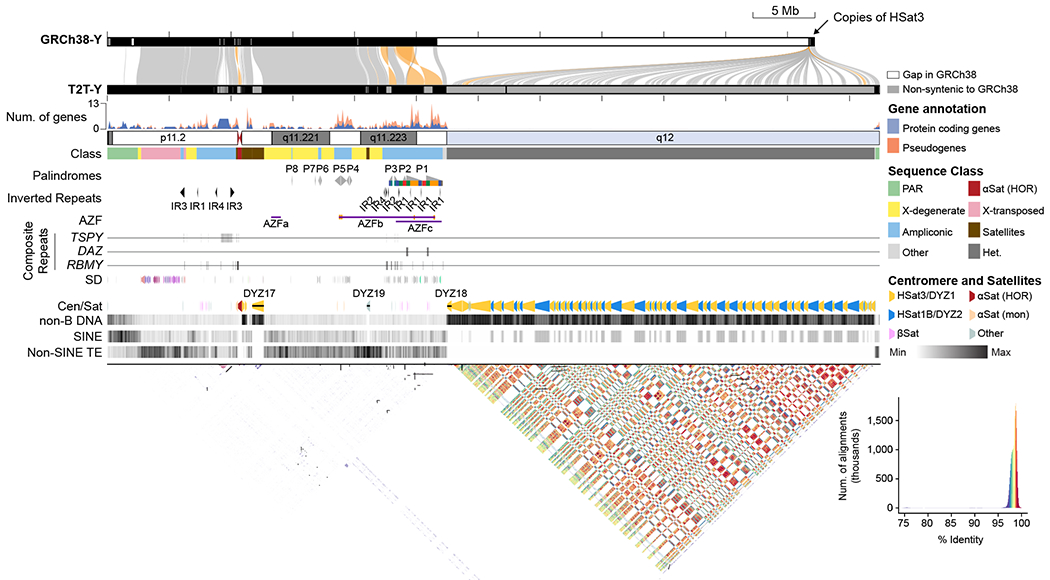
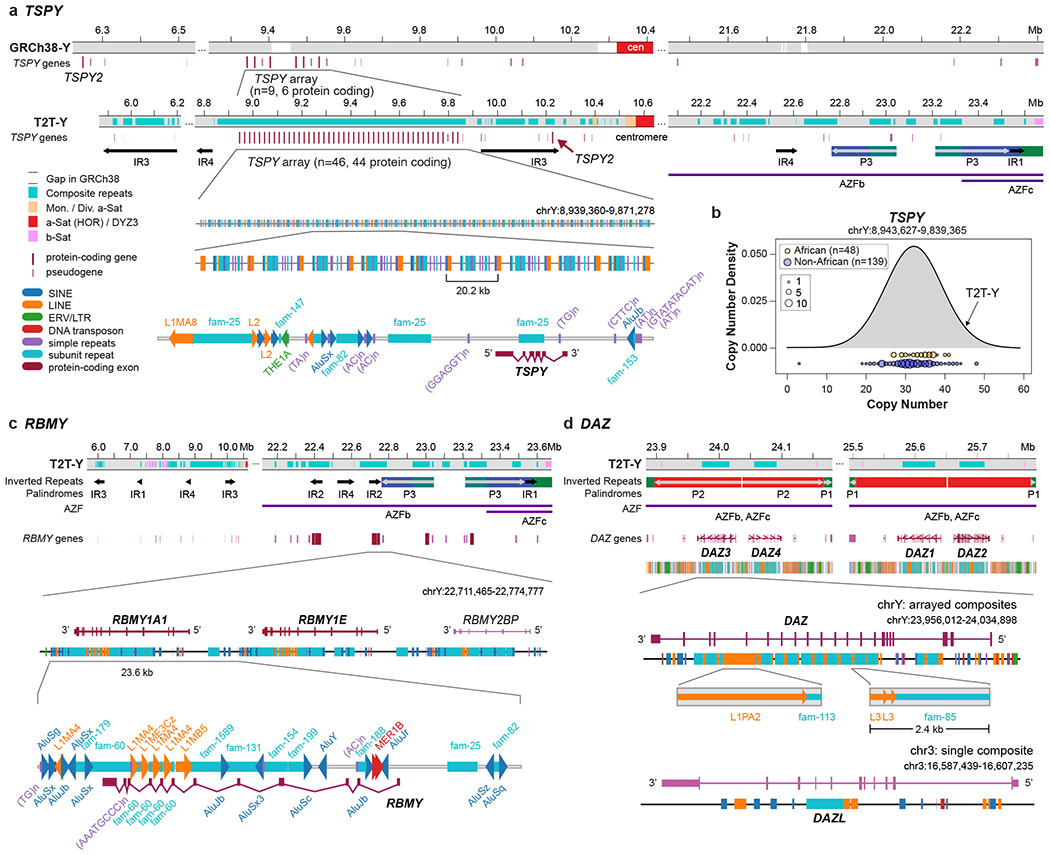
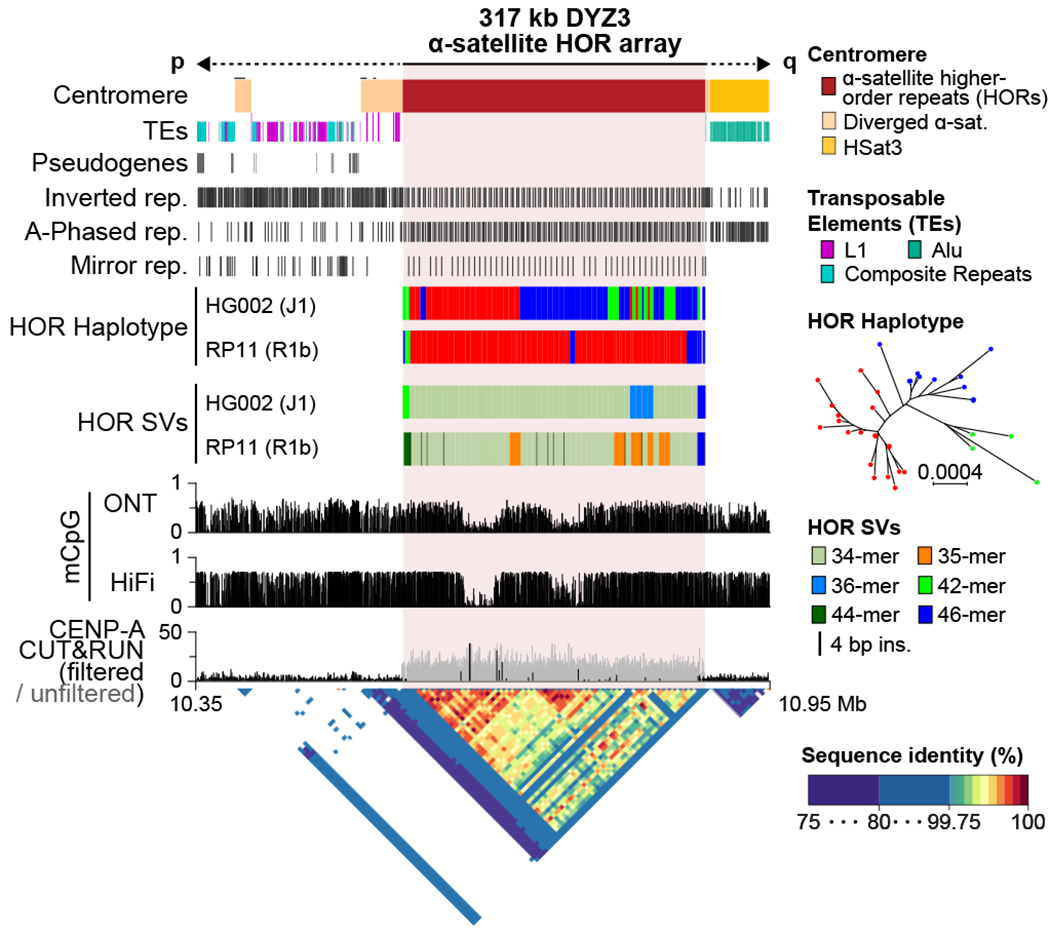
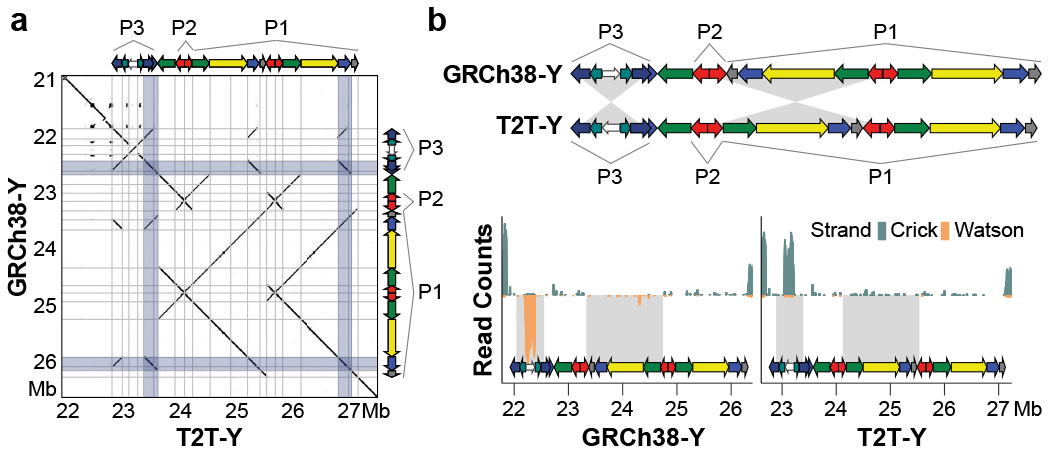
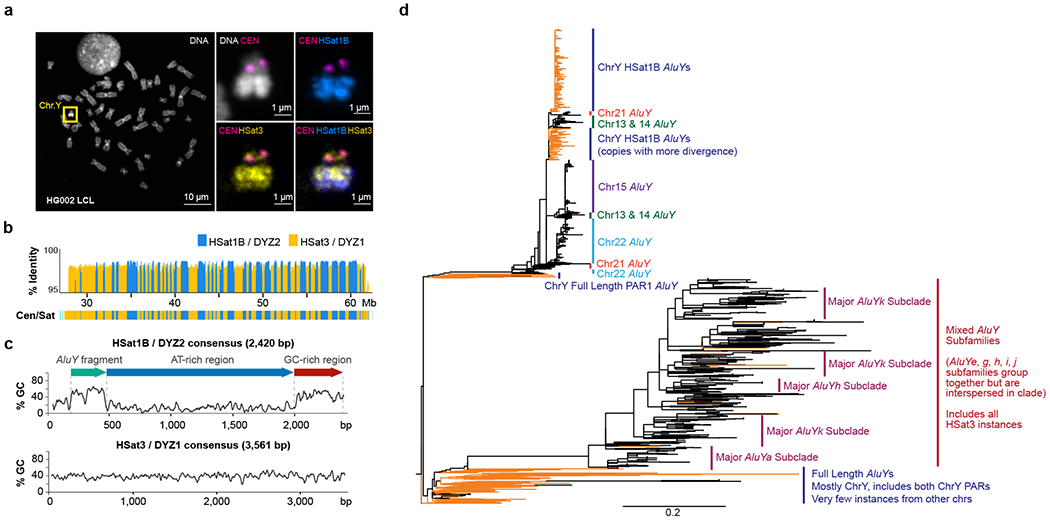
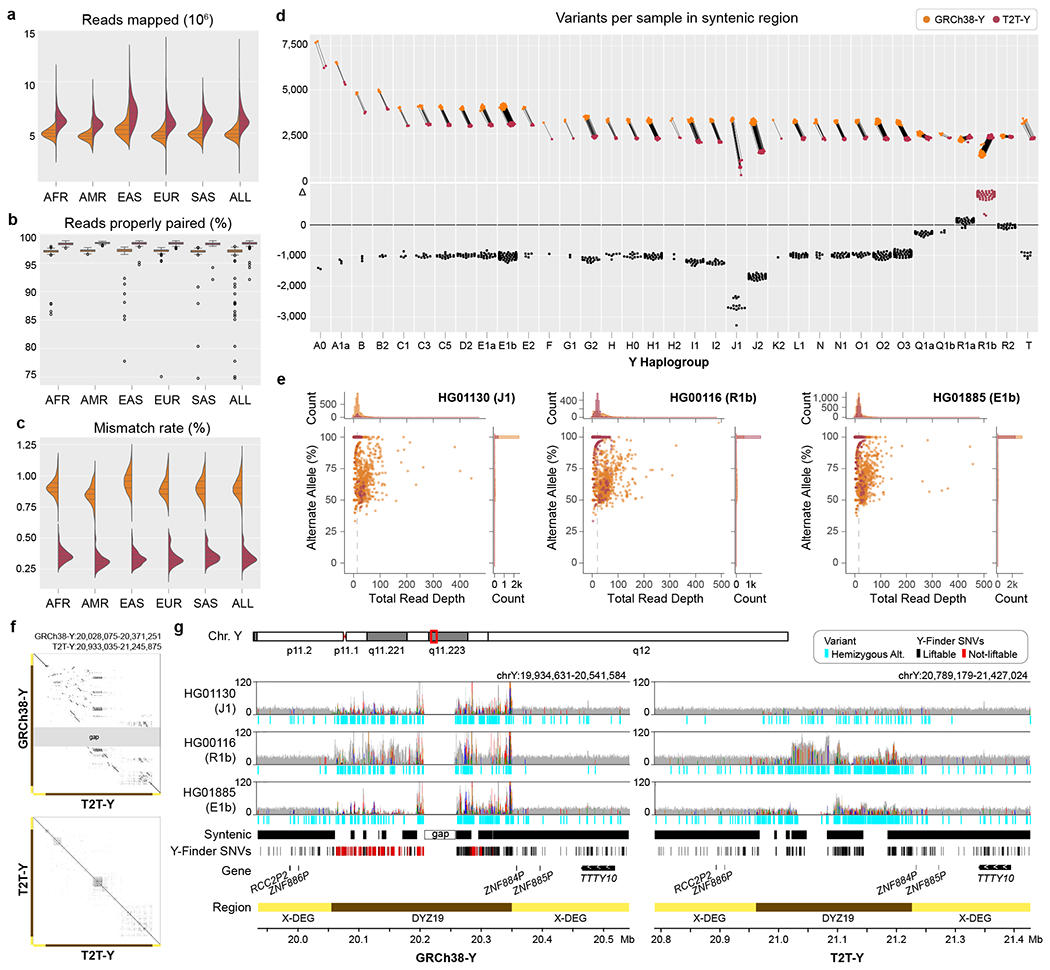
The human Y chromosome has been notoriously difficult to sequence and assemble because of its complex repeat structure that includes long palindromes, tandem repeats and segmental duplications1-3. As a result, more than half of the Y chromosome is missing from the GRCh38 reference sequence and it remains the last human chromosome to be finished4,5. Here, the Telomere-to-Telomere (T2T) consortium presents the complete 62,460,029-base-pair sequence of a human Y chromosome from the HG002 genome (T2T-Y) that corrects multiple errors in GRCh38-Y and adds over 30 million base pairs of sequence to the reference, showing the complete ampliconic structures of gene families TSPY, DAZ and RBMY; 41 additional protein-coding genes, mostly from the TSPY family; and an alternating pattern of human satellite 1 and 3 blocks in the heterochromatic Yq12 region. We have combined T2T-Y with a previous assembly of the CHM13 genome4 and mapped available population variation, clinical variants and functional genomics data to produce a complete and comprehensive reference sequence for all 24 human chromosomes.
© 2023. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
S.N. is now an employee of Oxford Nanopore Technologies; S.K. has received travel funds to speak at events hosted by Oxford Nanopore Technologies; A.F. is an employee of DNAnexus; C.-S.C. is an employee of GeneDX Holdings Corp.; N.-C.C. is an employee of Exai Bio; L.F.P. receives research support from Genetech; F.J.S. receives research support from Pacific Biosciences, Oxford Nanopore Technologies, Illumina, and Genetech; K.S. is an employee of Google LLC and owns Alphabet stock as part of the standard compensation package; W.T. has two patents (8,748,091 and 8,394,584) licensed to Oxford Nanopore Technologies; E.E.E. is a scientific advisory board member of Variant Bio, Inc. All other authors declare no competing interests.
Figures
References
-
- Skaletsky H et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- R35 GM124827/GM/NIGMS NIH HHS/United States
- R01 GM130691/GM/NIGMS NIH HHS/United States
- T32 GM007454/GM/NIGMS NIH HHS/United States
- UM1 HG010971/HG/NHGRI NIH HHS/United States
- R01 HG002939/HG/NHGRI NIH HHS/United States
- K99 GM147352/GM/NIGMS NIH HHS/United States
- R01 HG009190/HG/NHGRI NIH HHS/United States
- ZIA HG200398/ImNIH/Intramural NIH HHS/United States
- U01 HG011758/HG/NHGRI NIH HHS/United States
- R35 GM133747/GM/NIGMS NIH HHS/United States
- Z99 HG999999/ImNIH/Intramural NIH HHS/United States
- U24 HG010263/HG/NHGRI NIH HHS/United States
- R01 GM136684/GM/NIGMS NIH HHS/United States
- R01 HG010040/HG/NHGRI NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- R21 CA240199/CA/NCI NIH HHS/United States
- R01 CA266339/CA/NCI NIH HHS/United States
- R00 GM147352/GM/NIGMS NIH HHS/United States
- U41 HG006620/HG/NHGRI NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- U01 CA253481/CA/NCI NIH HHS/United States
- U24 HG007234/HG/NHGRI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U24 HG006620/HG/NHGRI NIH HHS/United States
- U24 HG010136/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R21 HG010548/HG/NHGRI NIH HHS/United States
- S10 OD028587/OD/NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- U01 DA047638/DA/NIDA NIH HHS/United States
- R01 GM123312/GM/NIGMS NIH HHS/United States
- R01 GM072264/GM/NIGMS NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
