

CRISPR/dCAS9-mediated DNA demethylation screen identifies functional epigenetic determinants of colorectal cancer
- PMID: 37612734
- PMCID: PMC10464368
- DOI: 10.1186/s13148-023-01546-1
CRISPR/dCAS9-mediated DNA demethylation screen identifies functional epigenetic determinants of colorectal cancer
Abstract
Background: Promoter hypermethylation of tumour suppressor genes is frequently observed during the malignant transformation of colorectal cancer (CRC). However, whether this epigenetic mechanism is functional in cancer or is a mere consequence of the carcinogenic process remains to be elucidated.
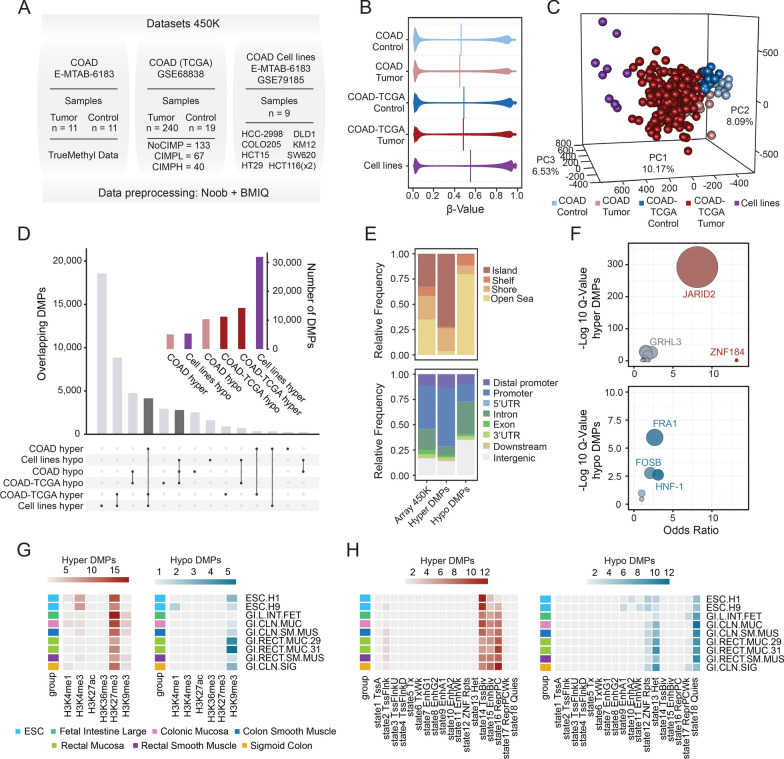
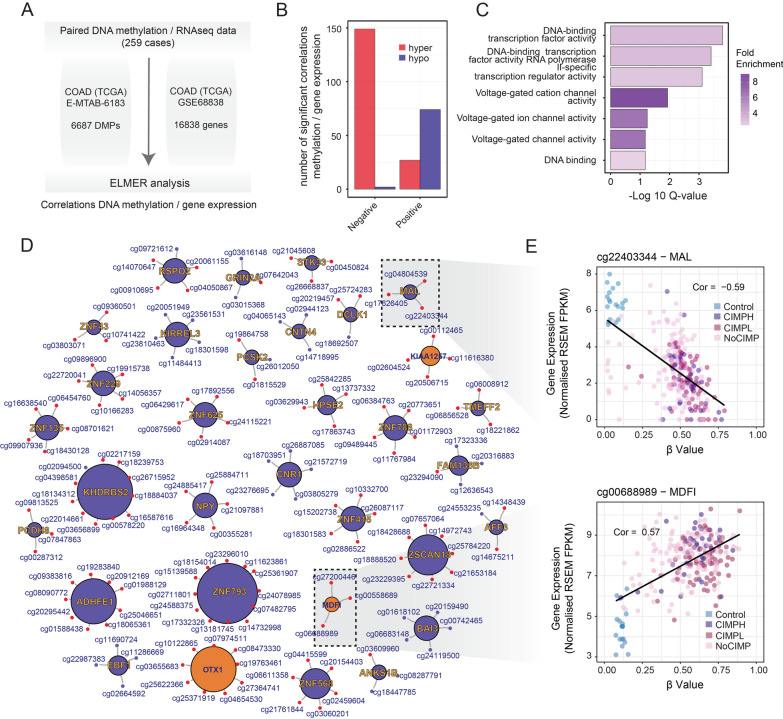
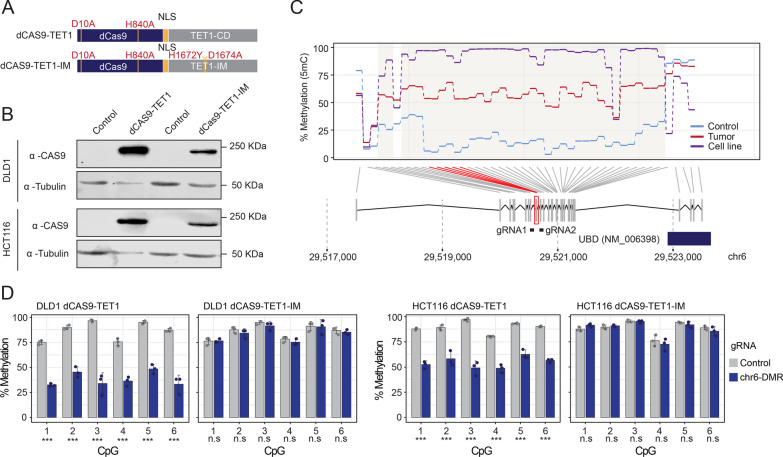
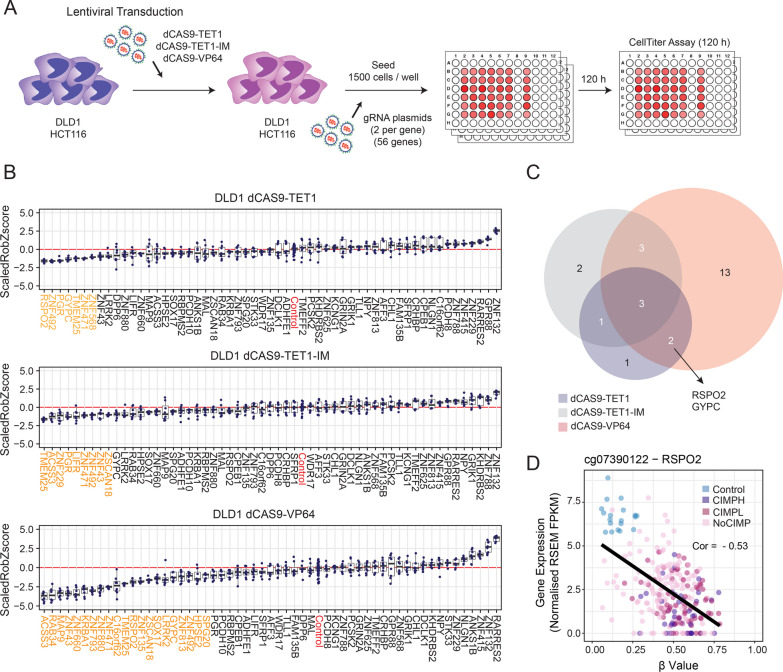
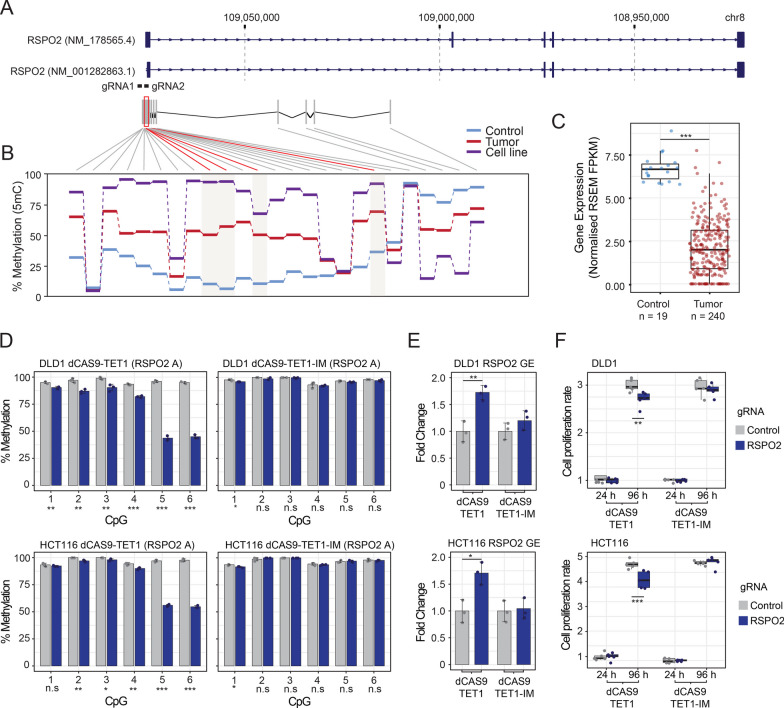
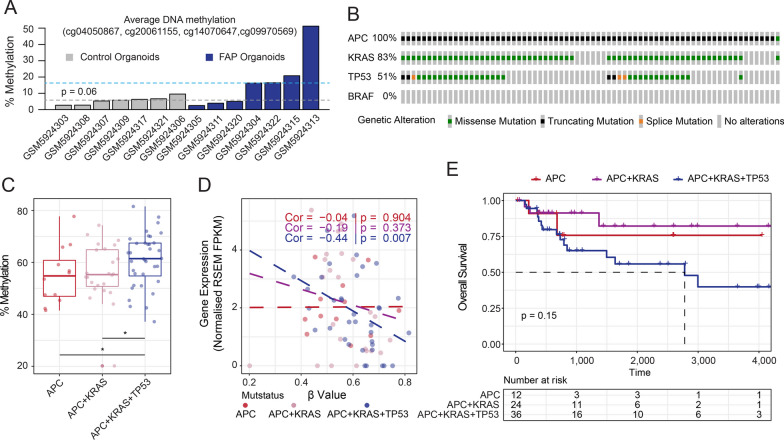
Results: In this work, we performed an integrative multi-omic approach to identify gene candidates with strong correlations between DNA methylation and gene expression in human CRC samples and a set of 8 colon cancer cell lines. As a proof of concept, we combined recent CRISPR-Cas9 epigenome editing tools (dCas9-TET1, dCas9-TET-IM) with a customized arrayed gRNA library to modulate the DNA methylation status of 56 promoters previously linked with strong epigenetic repression in CRC, and we monitored the potential functional consequences of this DNA methylation loss by means of a high-content cell proliferation screen. Overall, the epigenetic modulation of most of these DNA methylated regions had a mild impact on the reactivation of gene expression and on the viability of cancer cells. Interestingly, we found that epigenetic reactivation of RSPO2 in the tumour context was associated with a significant impairment in cell proliferation in p53-/- cancer cell lines, and further validation with human samples demonstrated that the epigenetic silencing of RSPO2 is a mid-late event in the adenoma to carcinoma sequence.
Conclusions: These results highlight the potential role of DNA methylation as a driver mechanism of CRC and paves the way for the identification of novel therapeutic windows based on the epigenetic reactivation of certain tumour suppressor genes.
Keywords: CRISPR screen; Colorectal cancer; DNA methylation; Epigenetics; Gene expression; Tumour suppressor gene.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–656. - PubMed
-
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. - PubMed
-
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
