

Prediction of mechanistic subtypes of Parkinson's using patient-derived stem cell models
- PMID: 37615030
- PMCID: PMC10442231
- DOI: 10.1038/s42256-023-00702-9
Prediction of mechanistic subtypes of Parkinson's using patient-derived stem cell models
Abstract
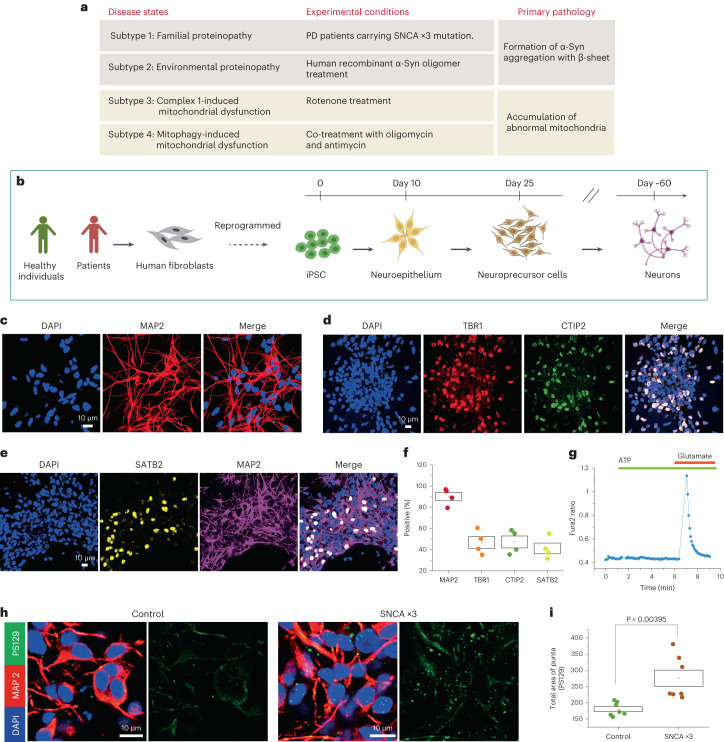
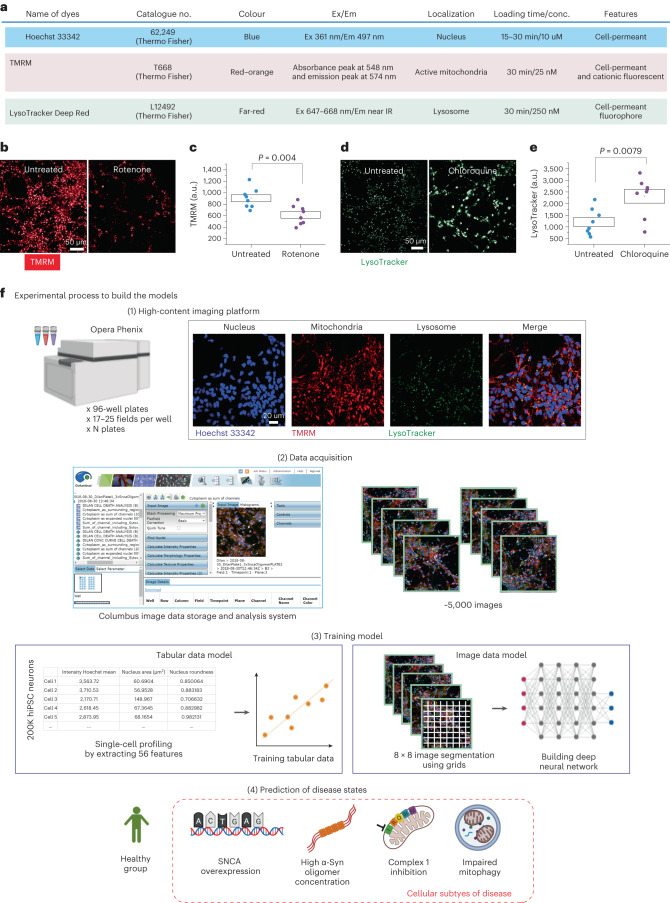
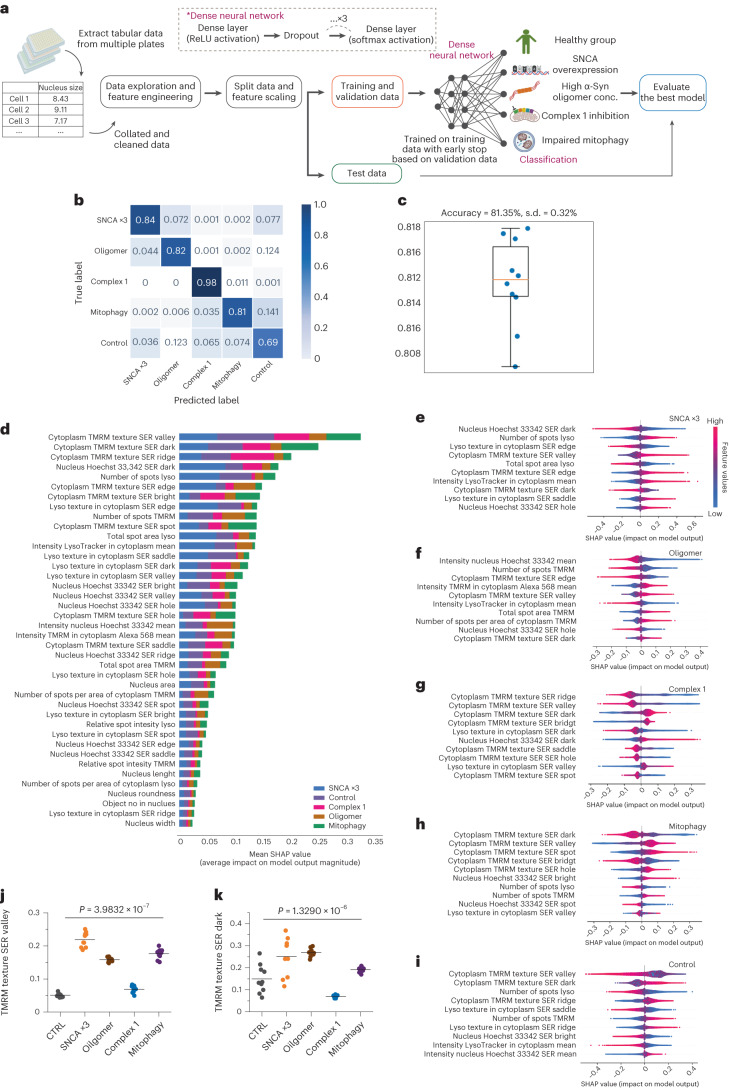
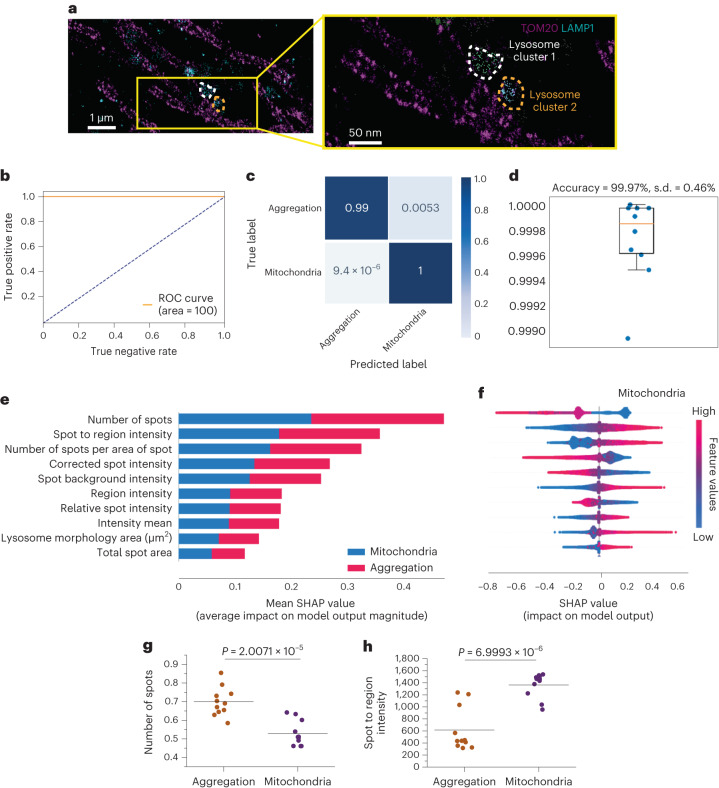
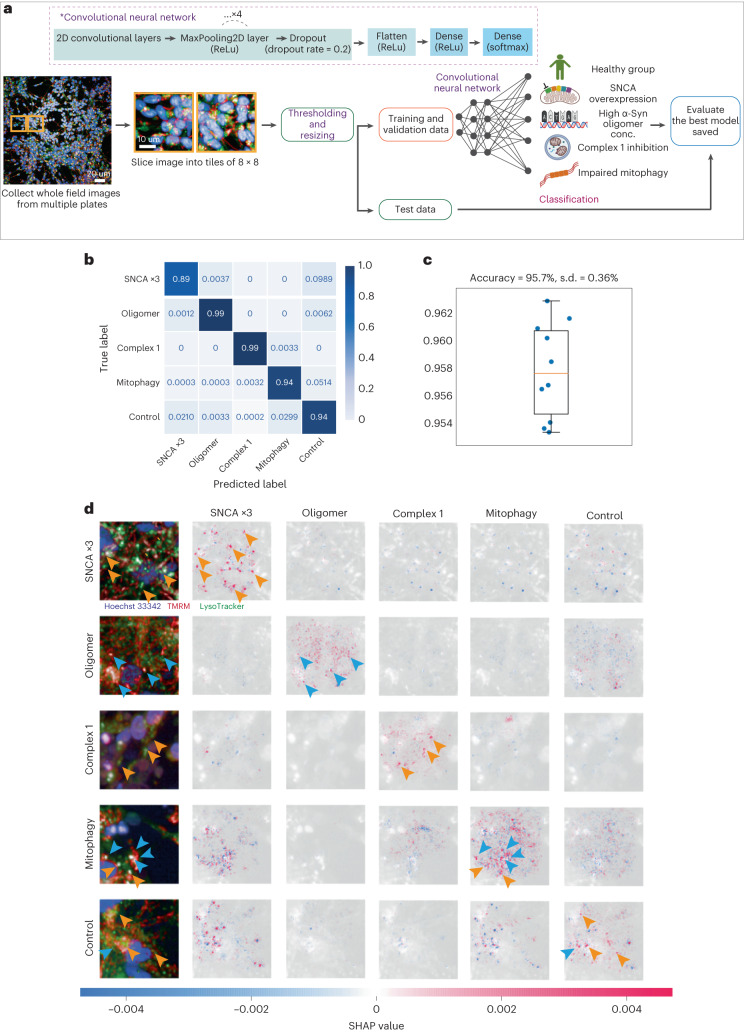
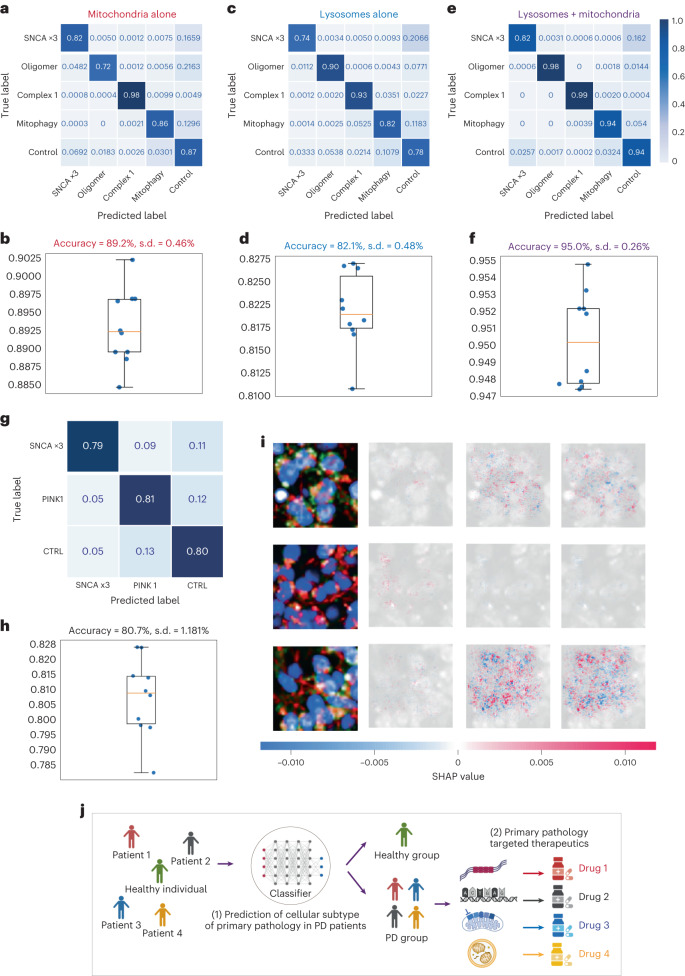
Parkinson's disease is a common, incurable neurodegenerative disorder that is clinically heterogeneous: it is likely that different cellular mechanisms drive the pathology in different individuals. So far it has not been possible to define the cellular mechanism underlying the neurodegenerative disease in life. We generated a machine learning-based model that can simultaneously predict the presence of disease and its primary mechanistic subtype in human neurons. We used stem cell technology to derive control or patient-derived neurons, and generated different disease subtypes through chemical induction or the presence of mutation. Multidimensional fluorescent labelling of organelles was performed in healthy control neurons and in four different disease subtypes, and both the quantitative single-cell fluorescence features and the images were used to independently train a series of classifiers to build deep neural networks. Quantitative cellular profile-based classifiers achieve an accuracy of 82%, whereas image-based deep neural networks predict control and four distinct disease subtypes with an accuracy of 95%. The machine learning-trained classifiers achieve their accuracy across all subtypes, using the organellar features of the mitochondria with the additional contribution of the lysosomes, confirming the biological importance of these pathways in Parkinson's. Altogether, we show that machine learning approaches applied to patient-derived cells are highly accurate at predicting disease subtypes, providing proof of concept that this approach may enable mechanistic stratification and precision medicine approaches in the future.
Keywords: High-throughput screening; Neurodegeneration.
© The Author(s) 2023.
Conflict of interest statement
Competing interestsThe authors declare no competing interests.
Figures








References
Grants and funding
LinkOut - more resources
Full Text Sources