Peroxisome disruption alters lipid metabolism and potentiates antitumor response with MAPK-targeted therapy in melanoma
- PMID: 37616051
- PMCID: PMC10575734
- DOI: 10.1172/JCI166644
Peroxisome disruption alters lipid metabolism and potentiates antitumor response with MAPK-targeted therapy in melanoma
Abstract
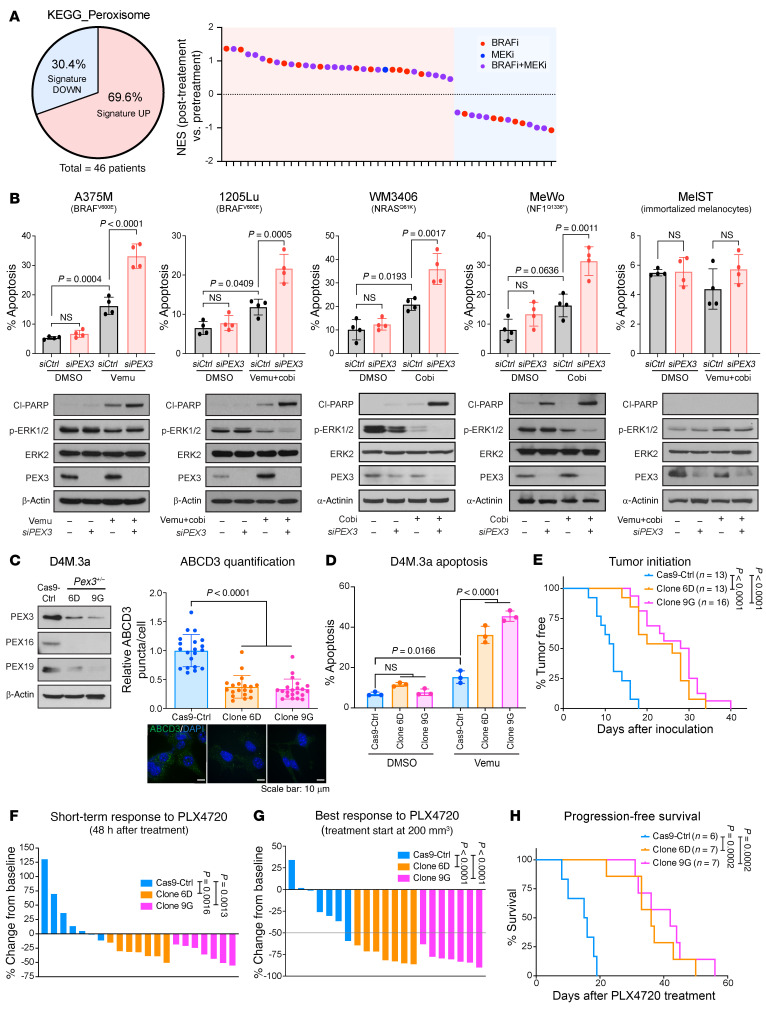
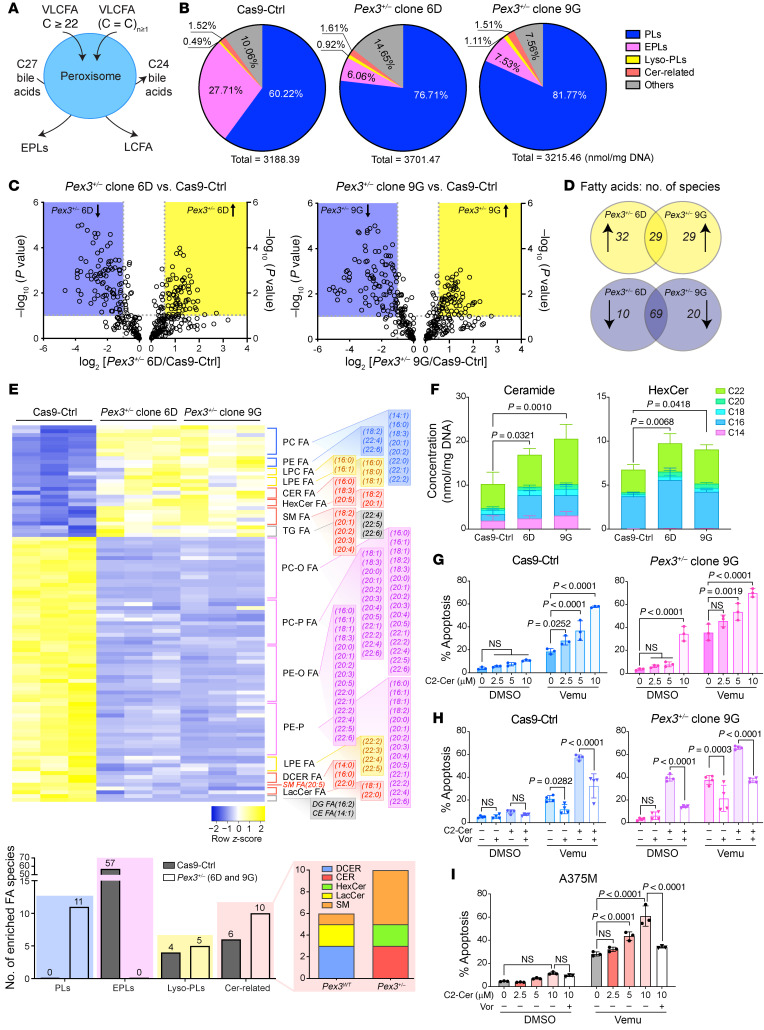
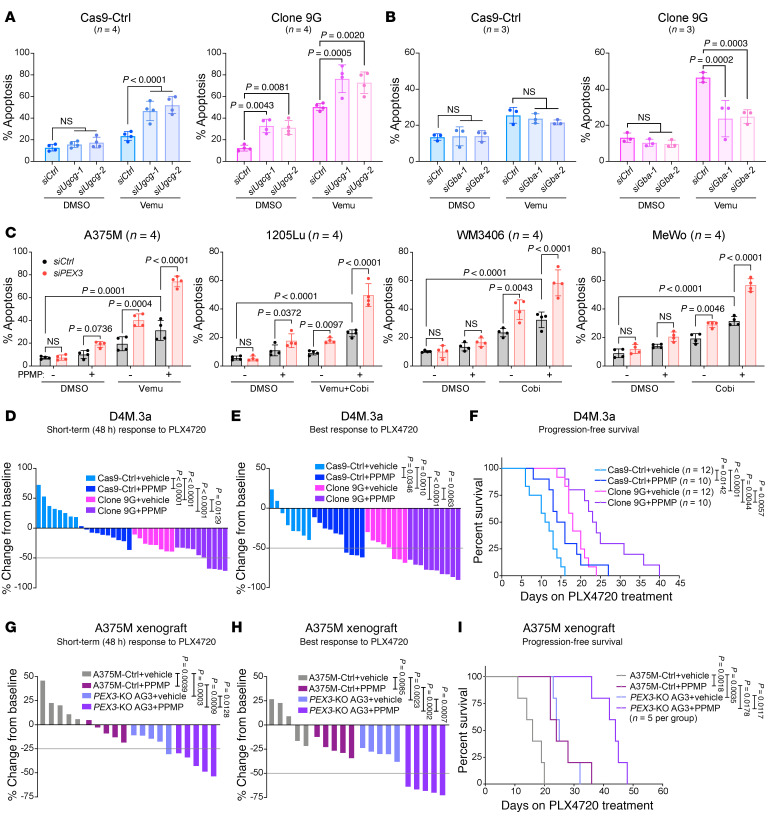
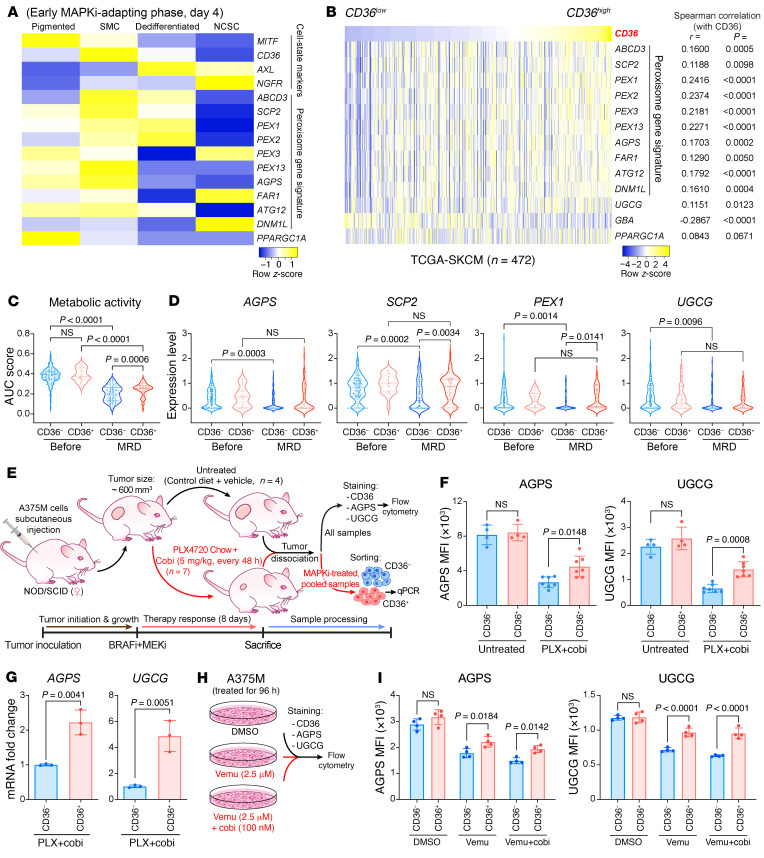
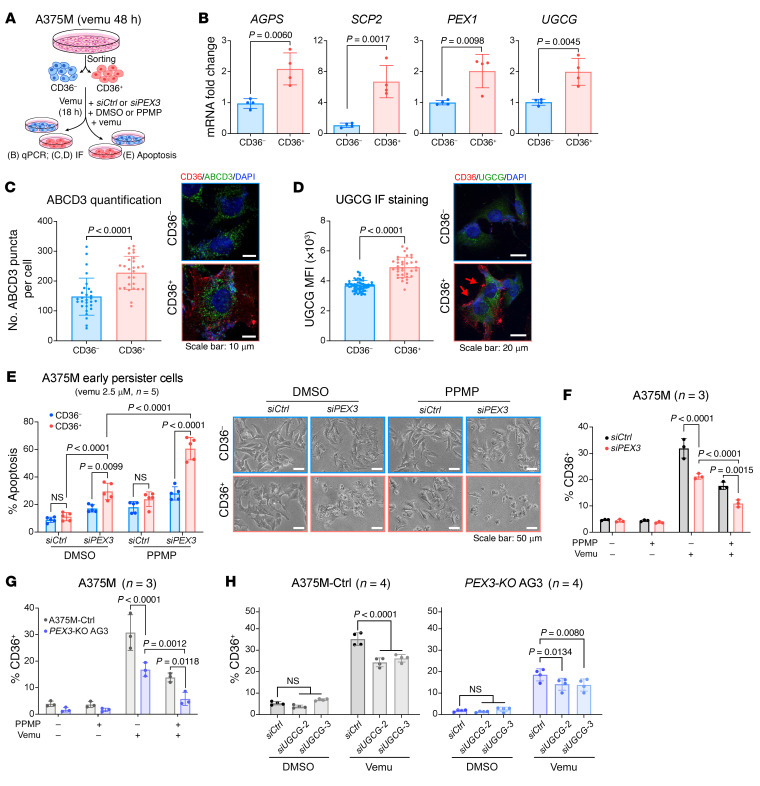
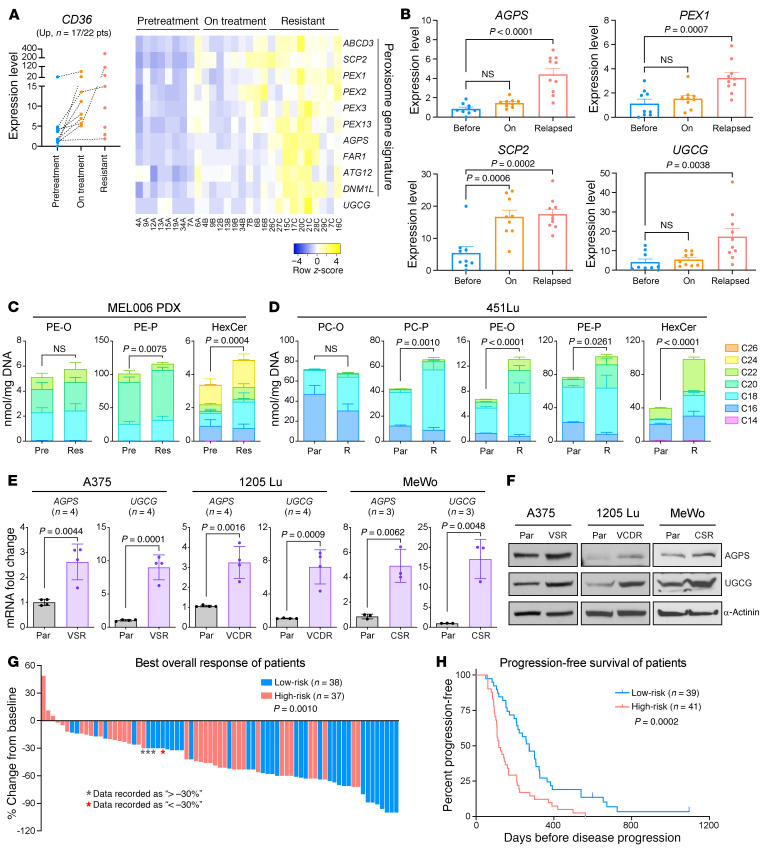
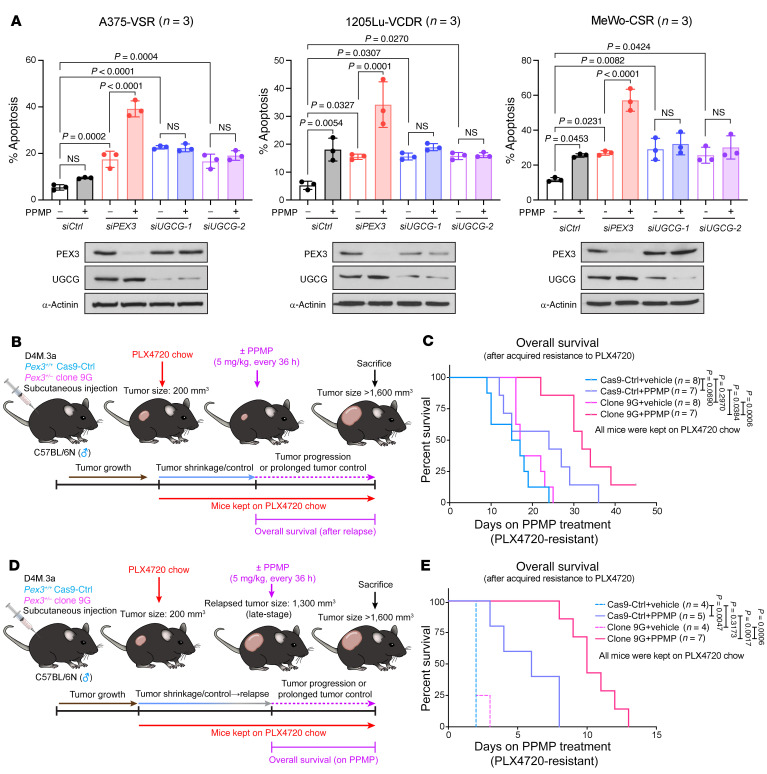
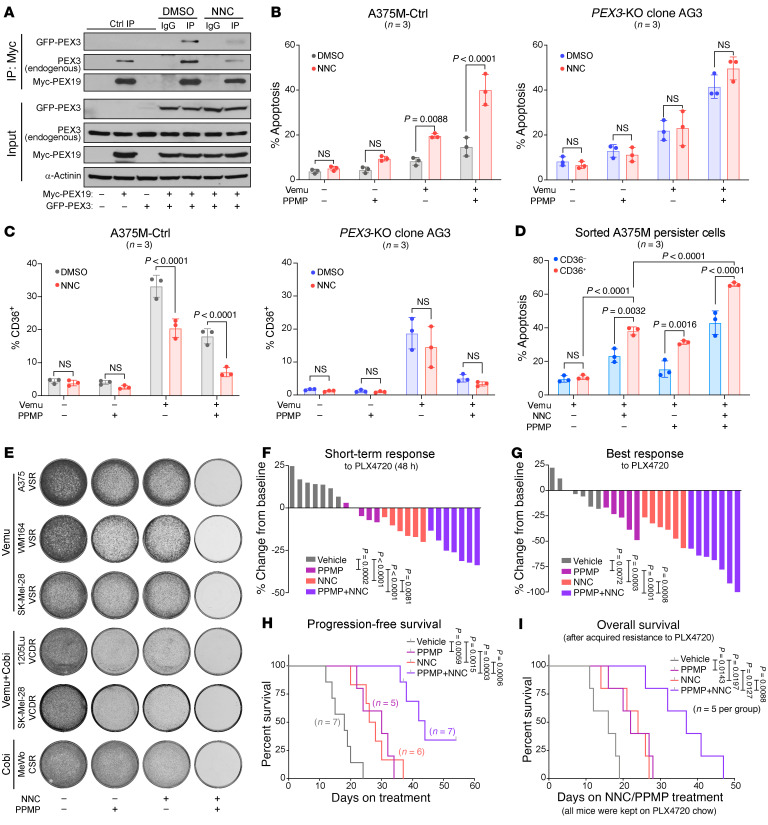
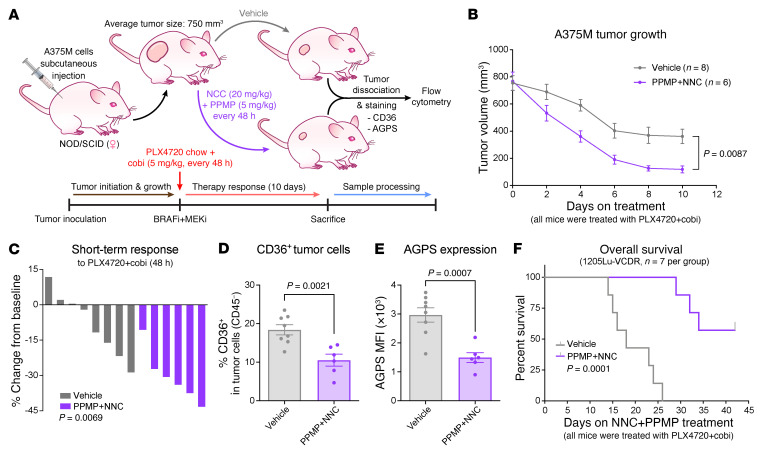
Melanomas reprogram their metabolism to rapidly adapt to therapy-induced stress conditions, allowing them to persist and ultimately develop resistance. We report that a subpopulation of melanoma cells tolerate MAPK pathway inhibitors (MAPKis) through a concerted metabolic reprogramming mediated by peroxisomes and UDP-glucose ceramide glycosyltransferase (UGCG). Compromising peroxisome biogenesis, by repressing PEX3 expression, potentiated the proapoptotic effects of MAPKis via an induction of ceramides, an effect limited by UGCG-mediated ceramide metabolism. Cotargeting PEX3 and UGCG selectively eliminated a subset of metabolically active, drug-tolerant CD36+ melanoma persister cells, thereby sensitizing melanoma to MAPKis and delaying resistance. Increased levels of peroxisomal genes and UGCG were found in patient-derived MAPKi-relapsed melanomas, and simultaneously inhibiting PEX3 and UGCG restored MAPKi sensitivity in multiple models of therapy resistance. Finally, combination therapy consisting of a newly identified inhibitor of the PEX3-PEX19 interaction, a UGCG inhibitor, and MAPKis demonstrated potent antitumor activity in preclinical melanoma models, thus representing a promising approach for melanoma treatment.
Keywords: Melanoma; Oncology.
Conflict of interest statement
Figures